
Abstract
The origin of nucleic acid template replication is a major unsolved problem in science. A novel stochastic model of nucleic acid chemistry was developed to allow rapid prototyping of chemical experiments designed to discover sufficient conditions for template replication. Experiments using the model brought to attention a robust property of nucleic acid template populations, the tendency for elongation to outcompete replication. Externally imposed denaturation-renaturation cycles did not reverse this tendency. For example, it has been proposed that fast tidal cycling could establish a TCR (tidal chain reaction) analogous to a PCR (polymerase chain reaction) acting on nucleic acid polymers, allowing their self-replication. However, elongating side-reactions that would have been prevented by the polymerase in the PCR still occurred in the simulation of the TCR. The same finding was found with temperature and monomer cycles. We propose that if cycling reactors are to allow template replication, oligonucleotide phenotypes that are capable of favorably altering the flux ratio between replication and elongation, for example, by facilitating sequence-specific cleavage within templates, are necessary; accordingly the minimal replicase ribozyme may have possessed restriction functionality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although in vitro selection of ribozymes (Cech 2002) lends credence to the idea of an RNA world (Gilbert 1986), we still have no replicase ribozyme (Johnston et al. 2001). Nonenzymatic synthesis of templates up to 55 nucleotides has been achieved on mineral surfaces (Ferris et al. 1996), but there is no replication, because templates do not recycle by unzipping (Kovac et al. 2003). Short oligonucleotide analogues can self-replicate (Von Kiedrowski 1986; Sievers and Von Kiedrowski 1994), but longer ones cannot because self-inhibition by strand association becomes prohibitive. Because long template replication would facilitate unlimited heredity (Szathmáry and Maynard Smith 1997), determining the conditions that could allow long nucleic acid replication to arise and be maintained would explain the details behind an early major transition in evolution (Maynard Smith and Szathmáry 1995).
Previous models of the origin of nucleic acid replication have consisted of ordinary differential equation models that incorporate high-level chemical assumptions (Kanavarioti and Bemasconi 1990; Kanavarioti 1994; Fernando and Di Paolo 2004) such as direct chain growth (Wattis and Coveney 1999), subexponential or parabolic template growth rates in a closed system (Von Kiedrowski 1993), ribozyme effects, or postulate idealized replication mechanisms (Wills et al. 1998). Macroscopic physical models of template replication, although ingeniously relaxing these assumptions, do not embody the order of magnitude differences in rates between phosphoamidate-bond (p-bond) and Watson-Crick base-pair (h-bond) events observed in nucleic acids, and have not yet demonstrated long template replication (Breivik 2001; Griffith et al. 2005). In order to integrate these diverse approaches, a low-level stochastic model of the underlying chemical kinetics of nucleotide and polymer dynamics was designed to explore the sufficient functional conditions for nonenzymatic long template replication.
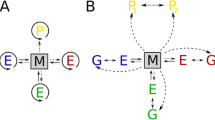
The details of the model and experiments to validate the model are described under Materials and Methods. The Results section presents experiments using the model to test the claim that cyclic reactor conditions, for example, those allowing denaturation-renaturation cycles, can facilitate nonenzymatic template replication. Lathe (2003, 2005) proposes that fast tidal cycling allows nucleic acids to associate and undergo ligation at high template and precursor concentration and high salinity when the tide goes out, and dissociate at low template and precursor concentration and low salinity when the tide comes in. Others have proposed that temperature oscillation in hydrothermal vent systems may serve a similar function (Kuhn 1972; Stein and Anderson 1984; Braun and Libchaber 2004). Alternatively, Gánti (1979, 2003) proposes that oscillation of monomer concentration in a metabolizing protocell can allow nonenzymatic template replication. All the experiments using the stochastic model show that this general class of solution is susceptible to an “elongation catastrophe” as shown in Fig. 1, in which Lathe’s proposed mechanism is compared to the behavior observed in the stochastic model. The principal aim of this paper is to give insight into the reaction mechanisms that take place in cycling reactors containing nucleic acid templates in the absence of enzymes.

Top: Adapted from Lathe (2005). Lathe proposes that salt increase on drying promotes duplex formation and monomer alignment for polymerization; salt removal on dilution then drives dissociation. Bottom: Results from the stochastic model reveal that perfect duplexes are only rarely formed, given random sequences of nucleotides. Instead staggered duplexes form and are then elongated by monomer attachment and by ligation to other staggered duplexes, resulting in “trapping” of sequence motifs and de novo synthesis of new sequences. Furthermore, because phosphoramidate bond formation is orders of magnitude slower than base-pair formation, premature detachment of dimers and trimers is overwhelmingly the most likely outcome compared to complete strand replication (shown by Lathe 2005).
Irrespective of the period of tidal cycling (Verga et al. 2006), the tidal chain reaction (TCR) differs from the polymerase chain reaction (PCR) in that there is no mechanism (e.g., a DNA/RNA polymerase complex) for preventing elongation side-reactions. Temperature cycling to obtain denaturation-renaturation cycles is functionally equivalent to tidal cycling with respect to elongation rate but is also more likely to cause strand breakage. Monomer concentration cycling does not facilitate template replication because at high monomer concentrations, there is elongation of strands at staggered ends and production of incompletely replicated dimers and trimers, and at low monomer concentrations, oligomers are ligated together. In all cases, the continued nonenzymatic formation of new nucleic acid polymers requires high monomer and template concentrations (in the millimolar range). These conclusions support the notion that protocells may have preceded template replication (Szathmáry et al. 2005). To conclude, we propose a possible solution to avoid the elongation catastrophe.
Materials and Methods
An informal description of the algorithm is provided, followed by a formal specification. Figure 2 shows the overall organization of the simulation. The simulation used a two-timescale technique to deal with the order of magnitude difference in characteristic times between (Watson-Crick base-pair) h-bond events and (phosphoramidate) p-bond events. An “event” means the formation or breakage of a bond or a bond type. Constrained by high computational costs, a very small volumeFootnote 1 was modeled as a flow reactor containing initially 20 random sequence complementary dimer or 4-mer double strands, giving an effective oligomer double strand concentration of 2 mM. Monomer concentration was held constant at either 0.8 or 2 mM. Using a variant of the Gillespie algorithm, the hydrogen-bond dynamics of the system were simulated until a quasi-steady state was reached, e.g., for a period of 0.0001–0.05 s.Footnote 2 Figure 4 describes the reaction rules used to calculate propensities. Once a steady state in the fast h-bond dynamics was reached, microstates from this steady state were sampled at fixed time intervals of 0.05 s. Each microstate had an associated propensity of p-bond formation, given by the sum of the number of potential p-bond forming configurations contained in that microstate multiplied by the rate of p-bond formation associated with each p-bond forming configuration. Also, each microstate had an associated p-bond breakage propensity, which was assumed to be simply proportional to the number of p-bonds in that microstate, therefore this value was equal across all microstates. A time was generated at which the next p-bond formation event was expected to occur, by entering the p-bond formation propensity into Eq. (1):
Similarly, an expected time was generated for the next p-bond breakage event. The event occurring first was executed. Roulette wheel selection was used to randomly choose a microstate in which to execute the event, with microstate weighting determined by the p-bond event propensity in that microstate. Once a microstate had been chosen, roulette wheel selection was applied over all p-bond events of the appropriate type in that microstate. In general, a time to the next event was always obtained by applying Eq. (1) to the propensity of that event. The event occurring earliest was the one chosen for execution. The particular bond that was chosen to undergo an event of the chosen class was obtained by percolation through a chain of roulette wheel selections, biased by propensities at each level in the hierarchy.

A high-level diagram of the overall simulation algorithm. Only a small sample of simulated templates, and only C and G nucleotides, are depicted.
Figure 3 illustrates the representation of nucleic acids used in the simulation. A polymer is defined as a contiguous molecule joined by h-bonds and p-bonds. Each polymer was represented on a separate unbounded grid. Each grid represented individual A, C, G, and T nucleotides on its vertices, hydrogen (h-bonds) on the vertical edges, and covalent bonds (p-bonds) on the horizontal edges. Thus, a large set of polymer secondary structures could be represented on the grid. Hairpins were excluded because p-bonds were confined to the horizontal edges. First, using this representation, a composite propensity of reaction was calculated for each polymer: the intrapolymer reaction propensity (IPRP). The IPRP was defined as the rate of reactions of type i, multiplied by the number of configurations that could undergo reaction type i, within that polymer at time t (excluding p-bond reactions). Figure 4 shows the reaction rules used to calculate the IPRP. Second, a propensity of collision was calculated for all possible reactions between polymers: polymer-polymer propensity (PPP). This was the propensity that the next event would be a collision event rather than an intrapolymer reaction. We assumed a well-stirred reactor, i.e., the probability of collision of all polymer pairs was equal. Third, the propensity that the next event would be a reaction between a monomer and a polymer was determined, so defining the monomer-polymer propensity (MPP). Having determined all composite event propensities, times were generated for each composite event type. The event type that occurred first was chosen for execution. Roulette wheel selection was used to percolate the event type decision to a particular location. If the earliest event was an intrapolymer event, then roulette wheel selection was used to choose which particular polymer would undergo the event. Roulette wheel selection was again applied to all possible events within that polymer, and an event was executed at time t+x, where x is the time generated using IPRP as the propensity. If the earliest event was a collision event, then the collision algorithm was executed, whereby two polymers where randomly chosen to undergo a collision, at two randomly chosen sites. A collision was deemed legitimate if it did not result in overlapping nucleotides or bonds; and if legitimate, an h-bond was formed between these two sites at time t + x, where x is given by applying PPP to Eq. (1), and the two polymers were joined to form one polymer. If the collision was not successful, time was updated by x, as above, but no change was made to the polymers. Similarly if the earliest event was a monomer-polymer association, roulette wheel selection was again used to choose a polymer onto which a monomer would attach, and again, roulette wheel selection was used to determine a site on that polymer onto which a monomer would attach. A monomer was attached by an h-bond at time t + x, where x is given by applying MPP to Eq. (1).

Top: The representation of polymers allows a very simple secondary structure that forbids hairpins. Therefore, any mechanism of replication requiring hairpins will not be possible in this model. Top left: Internal loops are possible. Top right: Breathing ends are possible. Bottom: Grid A shows a complementary double strand of length 5. Grid B shows a complementary double strand of length 10. Grid C shows a complementary double strand where the rightmost h-bond has been broken. Grid D shows a single strand of length 5. Grid E shows one of the possible polymers that may result when D collides with C. Grid F shows a polymer that may result when D collides with another single strand. This will be unstable because the h-bond is between a noncomplementary pair of nucleotides. Any polymer configuration is permitted by the model, as long as two steric constraints are obeyed: nucleotides cannot overlap each other, i.e., only a single p-bond and h-bond can occupy the same edge; one nucleotide can only be attached to one other nucleotide by an h-bond. The stability of a newly formed polymer is an emergent property of the strength of the stacking and h-bond forces that result immediately after association.

The above rules define all possible reactions within polymers (rules 1–5) between polymers (rule 7), and between polymers and monomers (rule 6). Each reaction is associated with a propensity, and this is used to calculate the IPRP, PPP, and MPP composite propensities used in algorithm 1.
The algorithms described above are defined formally below. Algorithm 1 describes how fast h-bond dynamics were run to a quasi-steady state between each p-bond event, and algorithm 2 describes the process shown in Fig. 2. A full annotation of the algorithms is available in the supplementary material.
Figure 4 describes how the IRPR, PPP, and MPP values were calculated using realistic reaction kinetics and free energies obtained from empirical studies (Cantor and Schimmel 1980; Turner 2000; Reynaldo et al. 2000; Rohatgi et al. 1996; Schoneborn et al. 2001; SantaLucia 1998; Xia et al. 1998). The p-bond formation and breakage rates were modeled on phosphoramidate bonds, although due to the timescale separation between h-bond and p-bond dynamics, the same overall behavior is expected irrespective of the absolute values of p-bond formation and breakage rates (see supplementary material). The probabilities of bond breakage and formation are functions of the local neighborhood configuration of bonds, monomer concentration, and temperature. The rules shown in Fig. 4 were applied to each bond or potential bond site on each polymer to calculate the rate of that bond breaking or forming and then used to calculate the composite propensities, IPRP, PPP, and MPP.
Rule 1: h-bond breakage rate, \( d_{m} = A_{m} e^{(E_{n} - Kn)/ RT} \). GC single h-bond breakage activation energy E a = 31.0 kJ. AT single h-bond breakage activation energy E a = 29.5kJ. Mean E a for noncomplementary h-bonds = 18.4 kJ. This resulted in approximately 10% mispaired nucleotides. Modifications applied due to stacking effects are described in Fig. 5. T is the temperature in Kelvin (either 275 or 300 K in these experiments). R = 8.314 × 10−3 kJ K−1 mol−1. The central h-bond in configuration B1 contains no h-bonds adjacent to it, and so it has the highest probability of breakage. The central h-bond in the BN configuration has four adjacent h-bonds, and so has the lowest probability of breakage. The ordering of stabilities of the central h-bond from least to most stable is B1 > B12 > B2 > B23 > BN1 > BN.Footnote 3 The Arrhenius constants for each neighborhood configuration are approximated by AB1 = 3.6 × 1013, AB12 = 1.8 × 1013, AB2 = 7.3 × 1012, AB23 = 1.8 × 1012, AB3 = 3.6 × 1011, ABN1 = 3.6 × 1010, and ABN = 1.8 × 1010. The entropy term K n was required to obtain the proper melting temperature curve for long strands (see supplementary material). K was empirically set to 0.01, n is the number of nucleotides not attached by h-bonds to another nucleotide, e.g., those at the dangling ends.

According to the nearest-neighbor (NN) and pyrimidine-purine (RY) stacking models, the local configuration of intra- and interstrand nucleotides influences the stability of h-bonds. To calculate the propensity of breakage of an h-bond, one has to check which instances of the above template configurations apply. The figure shows all the possible stacking rules that may be applied to the central h-bond between a 5′ G (blue square) and a 3′ C (red circle) nucleotide, labeled with a green arrow, given the appropriate nucleotide configuration. The 5′–3′ strand is shown at the top of each duplex. To see how to apply the rules to an h-bond between a 3′ C and a 5′ G, simply rotate the picture by 180o about the plane of the page, i.e., look at it from the back. From top left to bottom right: the GC dinucleotide core is assumed to result from two intrastrand stacks having total energy 2 × iCG. The GG and CC cores result from two interstrand stacks between C and G (with total energy 2 × eCG). The CC core results from one interstrand stack between G and G (with energy eGG). The additional effects of stacking at longer distances are modeled by the simultaneous application of four further rules, contributing stacking stabilization of the central h-bond at strengths +CGC, +CGG, and +CGGC, applied according to the satisfaction of the templates above; i.e., if the h-bond falls within the red highlighted region, then that h-bond receives the stacking stabilization associated with that region. With the inclusion of A and T nucleotides, additional configurations must be considered. The stabilities have numerical values as follows (kJ): iGC = 1.12, eCG = 0.92, eGG = 2.17, iAT = 0.44, eTA = 0.5, eAA = 0.58, iGT = 0.72, iAC = 0.72, eTG = 1.28, eCA = 1.30, eAG = 1.45, CGC = 1/5, CGG = –4/5, and CGGC = 2/5. A scaling factor of +1.8 was typically used to multiply all these stacking effects, and was adjusted to allow approximation of experimentally observed T m values. The effectiveness of these adjustments can be seen by looking at the observed vs modeled T m ’s for various oligonucleotide sequences in Fig. 7.
Rule 2: Catalyzed h-bond formation rate = 106 s−1 adjacent to other h-bonds.
Rule 3: Zipper h-bond formation rate = 106 s−1 irrespective of the distance from the potential h-bond to the closest nucleation site (a nucleation site is a pattern of three adjacent h-bonds). Note that the simultaneous application of rules 2 and 3 results in h-bonds being twice as likely to form next to another h-bond than anywhere else along a duplex.
Rule 4: Template directed p-bond formation rate = \( p\_form\_rate = 0.6e^{{-25} / RT} \).
Rule 5: p-bond breakage rate = \( rate\_p\_break = 1.32 \times 10^{12}\,e^{{-110}/ RT} \), n.b p-bond breakage rate exceeds p-bond formation rate, above 350 K (see supplementary material)l. Note that rules 4 and 5 are only applied in the outer loop of algorithm 2, not in algorithm 1.
Rule 6: Monomer attachment. Complementary rate = 106 s−1. Noncomplementary rate = 103 s−1. Stacked monomer attachment (i.e., attachment of a monomer next to an h-bond) is 100 times these values.
Rule 7: Collision rate between any two polymers = 5 × 109 s−1 M−1. Note that this is a bimolecular rate constant because it is multiplied by two concentrations. The total collision propensity is given by (5 × 109) × (no. of polymers/N A V), where N A = Avogadro’s number, and V is the volume. We set N A V = 104 when the tide was out and N A V = 105 when the tide was in. When a collision occurred, the two random nucleotides between which a new h-bond (or transient “stacking” s-bond; see Fig. 6) would form were chosen, and the polymers were combined at that site. If the combination resulted in no overlap of nucleotides on the two-dimensional (2D) grid, then the collision was successful and polymers were combined; otherwise, the polymers remained separate. “Rule” X: Spontaneous p-bond formation by association of monomers was forbidden because the rate was too low to be significant at the timescales considered. However, spontaneous p-bond formation between oligomers by single-stranded and double-stranded (nontemplated) end ligation was allowed; see Fig. 6.

Stacking s-bond formation (shown as a gray bond) occurs between blunt ends, with a certain probability whenever two polymers collide. An s-bond-mediated collision occurs if, upon collision, a random number between 0 and 1 is greater than 0.3 + 0.25 × log(number of free h-bond sites on polymer A × number of free h-bond sites on polymer B) and both strands have fewer than eight p-bonds each. The mean rate of s-bond breakage has the same profile as the rate of h-bond breakage in the B3 configuration and is adjusted by stacking effects as follows. s-break rate = AB3 e −(Es + stack). AB3 = 3.6 × 1011 is just the Arrhenius constant for the intermediate h-bond breakage configuration, Es = 15.5kJ (an overestimate necessary to observe s-bonds in the small number of templates simulated), and stack = 3.0 × the NN stacking energy contribution due to application of the templates in Fig. 5. There is a certain rate of p-bond formation on s-bonds, which is assumed to be the same as the rate of templated p-bond formation, i.e., \( p\_form\_rate = 0.6e^{{- 25}/ RT} \). In practice the net ratio of spontaneous-to-template p-bond formation will depend on the specific chemical system.
Figure 5 describes how some more elaborate calculations of interstrand and intrastrand stacking forces were implemented (SantaLucia 1998; Cruz et al. 1982) because this was known to influence the tendency for elongation and contributes to “phenotypic” diversity (Sinclair et al. 1984; Zielinski and Orgel 1987a, b, 1989), for example, allowing GCGC to replicate but tending to cause CGCG to elongate.
Finally, transient “stacking” bonds were introduced, capable of mediating blunt end ligation of templates; see Fig. 6. This was necessary to match recent results observed experimentally in real chemical systems of pure dimers capable of elongation despite the fact that templated p-bond formation could not have occurred due to their extremely low melting temperatures.
To check that the above parameters produced the correct behavior, control experiments were conducted to calculate the melting temperature curve for AnTn and GnCn oligomers; see Fig. 7. To check h-bond kinetics, dissociation behavior from double-stranded to single-stranded states was measured as a function of strand length and temperature (Fig. 8). To check p-bond kinetics, an experiment was conducted in a reactor closed to mass and initialized with only GC dimers, at 275 K (Fig. 9). See the supplementary material for a control showing the different behaviors of GCGC and CGCG oligomers.

Melting temperatures (T m ) for AnTn and GnCn oligomers at concentration 20 mM. N = length. The x-axis shows 1/N and the y-axis shows 1/T m . Circles show the points at which T m was measured. The outlier is a 6-mer of sequence AAGCTT, which, as expected, shows an intermediate T m between A6T6 and G6C6.

The rate of dissociation of ds-GCGC 4-mers at different temperatures is as expected, denaturation being more rapid at higher temperatures. Initial concentration: 10 mM double-strands. Note that GCGCG takes longer to dissociate than GCGC at the same temperature and does not dissociate to as great an extent.

A batch reactor was initialized with 20 mM GC dimers at 275 K. The continuous lines show expected rates in a simple minimal replicator model for GCGC formation (increasing curve) from GC (decreasing curve) with the same p-bond formation kinetics (Von Kiedrowski 1993) as in the stochastic model. The ODE model assumes no elongation reactions beyond GCGC, whereas the stochastic model shows that such reactions are inevitable unless special blocking of ends is undertaken. Note that Zielinki and Orgel had to block template ends in their nonenzymatic replication experiments to prevent elongation (see supplementary material).
In order to check whether the model was capable of template replication in the presence of Q-beta-like sequence-independent RNA replicase ribozyme,Footnote 4 a model of this molecule was implemented as another “monomer” type present at a low copy number and capable of executing additional local bond formation and breakage rules. Figure 10 shows the intended mechanism of the particle, the local neighborhood rules that carry out this mechanism using the grid representation, and the replication and elongation behaviors observed from direct screenshots of the simulation.

Top: The Q-β like RNA replicase is shown as a purple square, attached to the p-bond at its base. The top strand is oriented 5′–3′. The ribozyme is only permitted to attach to a p-bond if there is no p-bond on its left (a). Procession can only take place if there is a p-bond at the top of the square, and so initially the ribozyme is nonprocessive (np). Here it waits, and stabilizes any h-bond that is formed within the purple square (blue circle). The h-bonds may be formed due to the attachment of either an oligomer or a monomer. In either case, the h-bond will be stabilized. The two h-bonds below the ribozyme are always destabilized (red crosses). Once two h-bonds are present in the purple square, the ribozyme catalyzes the ligation of the two nucleotides in its top corners (c). A p-bond is now present at the top of the square and procession can take place (p). The cycle starts again and continues until the ribozyme reaches the end of the template strand, at which point it dissociates. Bottom left: The above behavior is an emergent property of the application of the rules shown here. Grid A shows the configuration in which replicase attachment to a strand can occur. Grid C shows the configuration of catalytic effects exerted once the ribozyme is attached. Grid D shows the configuration in which detachment takes place. Grid P shows the configuration in which procession to the right (5′→3′) occurs. Bottom right: Some snapshots of replicase function. In the first three cases, a strand is being successfully replicated. In the bottom case, elongation is taking place, turning a staggered end into a blunt end. Thus, even the Q-β-like RNA replicase model tends to promote elongation at a low rate.
Tidal cycling was modeled as regular cyclic dilution and low salinity “spikes” sufficient to completely denature effectively all double strands. High tide (spike) dilution was assumed to reduce the concentration of strands by 10 times, and the effect of low tide salinity was to reduce the effective h-bond breakage activation energy by a constant value of 3 kJ per h-bond, so making h-bonds more unstable. The tide was applied at various periods as a square threshold function on a sine wave, i.e., high tide occurred when sin(t/period) > T, typically T = 0.9.
Two temperature cycling experiments were conducted. The first used spikes of 330 K over a baseline of 280 K, with very short high-temperature spikes producing complete denaturation. Spike length was defined as that sufficient to break p-bonds. The interspike interval was defined as the time required to make 100 p-bonds; s-bonds were not allowed in this experiment. In the second experiment, temperature spikes were administered in replacement of high tides to a value of 300 K from a baseline of 275 K; s-bonds were allowed. Experiments with fixed monomer concentrations and with monomer cycling were carried out with monomer concentration varying from 0.6mM to 60mM, with high monomer concentration replacing high tide events.
In summary, the major obstacles encountered in producing the model were (i) the vast number of distinct configurations that nucleic acid polymers could adopt transiently, resulting in many possible types of interactions; (ii) the order-of-magnitude difference in characteristic timescales between Watson-Crick base pairs (h-bonds) and phosphoramidate bonds (p-bonds); and (iii) the high interdependency of reaction rates, due to hydrogen bond stacking interactions, and interpolymer reactions. These problems were solved, respectively, by (i) limiting possible secondary structures to those without hairpins, and simulating very small volumes containing a few templates; (ii) using a relaxation method to allow h-bond dynamics to run approximately to equilibrium before each p-bond event; and (iii) using a very efficient stochastic algorithm (Gibson and Bruck 2000; Elf and Ehrenburg 2004).
Results
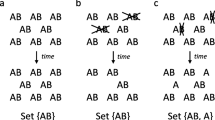
A reactor was initialized at 280 K with 20 double-stranded complementary 10-mers, each consisting of a random nucleotide sequence. This means that there is an initial concentration of 2 mM double stranded 10-mers or 4 mM single-stranded 10-mers in the reactor. In addition, identical and constant concentrations of A, C, G, and T monomers were provided at a total concentration of 2 mM. Visual inspection revealed the rapid template-mediated production of dimers (by ligation between monomers stacked on 10-mer single-stranded templates or on the exposed single-stranded parts of splint junctions). Simultaneously, strand elongation occurred by (i) ligation at splint junctions between staggered oligomers and (ii) ligation between monomers and staggered ends. Trimers began to form once the dimer concentration approached that of monomers. This is trivially triplet replication, because 3-mer motifs embedded in longer oligomers are serving as templates for ligation between monomers and dimers. However, there was an inexorable process of elongation. As the templates became longer, dissociation became less likely, and embedded motifs were therefore no longer capable of replication. Although there were rare instances of 4-mer replication and 5-mer replication, due to between-oligomer ligation on templates, these newly formed copies typically become embedded into the elongating strands and so were unable to undergo further rounds of replication. Thus, elongation side-reactions destroyed the capacity for motifs to continue to act as templates. Novel motifs were created at a rate higher than the rate of replication of old motifs, thus information in the form of long sequences was not heritable. The distribution of template lengths over the course of the experiment is shown in Fig. 11 (top left).

Top left: Length distribution over reaction events for a nontidal system initialized with 20 double-stranded 10-mer complementary random sequences and a constant supply of monomers of all types at a 2 mM total concentration. The total time of the experiment is 2 × 107 s. The maximum length strand at the end of the experiment is 35 p-bonds long. Dimers predominate the reactor. Bottom left: With tidal cycling at a period of 18,000 s, elongation is still observed. By 2.8 × 106 s, 24-mers are observed. Top right: Nontidal conditions with a low monomer concentration (0.8 mM). Elongation to 57-mers takes 6.6 × 107 s. Bottom right: Tidal conditions with a low monomer concentration (0.8 mM) results in elongation to 48-mers within 1.6 × 10 s.
Can tidal cycling help to remedy this problem? The claim by Lathe is that strand dissociation at high tide can free embedded motifs, allowing them to enter another round of replication; see Fig. 1. Tidal cycling was introduced with period 18,000 s, with dilution and reduced salinity lasting for 10% of each cycle, sufficient to allow complete dissociation of strands at high tide. The length distribution of templates under the influence of tidal cycling is shown in Fig. 11 (bottom). Under both high (left) and low (right) monomer concentrations, elongation was still observed with tidal cycling. Indeed the rate of elongation was approximately an order of magnitude faster with tidal cycling than without. This finding was not dependent on the period of cycling, salt concentration, or monomer concentration (see supplementary material), nor did it depend on the presence of s-bonds.Footnote 5 Direct visual inspection reveals that tidal cycling promotes elongation by splint junction ligation that occurs after reannealing of templates as the tide goes out. Experiments at higher temperatures (e.g., 320 K) revealed neither elongation nor replication; see Fig. 12. Temperature oscillation did not increase the capacity for replication, for reasons described in the legend to Fig. 13. Oscillation of monomer concentration did not increase the capacity for replication; see Fig. 14a. This is because at high monomer concentrations, oligomers were produced (Fig. 14b, bottom right) and later used to elongate templates at low monomer concentrations (Fig. 14b, top left).

A representation of reaction mechanisms directly observed at high and low temperatures. Left: Staggered association (A) and ligation at splint junctions (B) are the mechanism observed to produce elongation at low temperatures and upon renaturation. Right: At high temperatures, elongation does not occur due to premature strand separation (C), short strands outcompeting long-strand replication (D), and p-bond breakage (E).

a Elongation was observed with temperature oscillation. The graph shows time (arbitrary units) vs. length (p-bonds) against template frequency. An experiment was conducted with square wave temperature oscillation, consisting of a baseline of 280 K, with intermittent spikes of 330 K intended to cause long-strand denaturation. Three high-temperature spikes were administered over the course of the experiment. The s-bond dynamics were switched off. At 280 K the average time between p-bond formation events was 9 h, and breakage events were rare. At 330 K the average time between p-bond breakage events was 20 min, and p-bond formation events were rare. This made temperature oscillation a very precarious mechanism for denaturing strands, because p-bond breakage at the sort of temperatures required for long-strand denaturation is likely to destroy the long strands. The reaction chamber was designed such that a high-temperature spike occurred every 100 p-bond formation events, lasting for 2 p-bond breakage events. Only sequences up to 4 in length had been replicated after 3.13 × 108 s, with a system initialized with 20 random 5-mers. Temperature oscillation, although relieving product inhibition, does not allow long template replication, because splint junction-dependent elongation is just as much of a problem, and is equally benefited by reshuffling of strands when the temperature falls, as it is in the tidal cycling-mediated renaturation. The balance between elongation and replication is not shifted by the temperature oscillation procedure, ruling out the mechanisms proposed by Kuhn (1972) and Stein and Anderson (1984). b Temperature oscillation with s-bonds activated: range, 275 to 300 K, with 300 K “spikes” occurring according to the same function as high tides. Maximum length = 44; mean length = 7.9; p-bond events = 2074. Again, no replication of the initializing10-mers was observed.

a Monomer oscillation, as predicted, does not result in replication of 10-mers. Monomer concentration was oscillated from 0.8 to 2 mM, with 2 mM spikes occurring at the same frequency as tides. Maximum length = 34; mean length = 5.2; p-bond events = 1000. b The influence of monomer concentration on template elongation and replication. In no case was replication of a strand longer than six nucleotides in length detected. The x-axis shows strand length, the y-axis shows frequency, and the z-axis shows time. Top left: [Monomer] = 0, [initial polymer] = 0.00063 M. Templates elongate to a maximum of 95 in length. The experiment lasts for 109 seconds. At the end, there is only one complex 85 p-bonds in length. Top right: The control case. Trial duration = 3.3 × 108 s. Initial [4-mer] = 0.00063 M and [monomer] = 0.0006 M. Elongation occurs to 30-mers, with maximum replication length = 6. Bottom left: The polymer association rate constant was reduced by five times. This resulted in weaker association between polymers, but the same rate of monomer attachment, so reducing elongation to 23-mers, and allowing oligomers to dominate the length distribution, after 1.4 × 108 s. Bottom right: As above, except that the monomer concentration was increased 100-fold, from 0.0006 to 0.06 M. Dimer and trimer production dominated, after 29842.2 s.
Further checks for the presence of replication were carried out for the tidal cycling case. A brute force motif finding method was used to examine the distribution of all motifs in the reactor at the end of an experiment. How different is this distribution from a distribution that would be expected by random synthesis of sequences with the same length distribution and nucleotide composition as in the final reactor? A signature of replication is the clustering of sequences in sequence space. The number of sequences separated by a Hamming distance of <2 from another sequence would be expected to be greater in a reactor in which template replication was occurring, compared to a randomly generated set of sequences. One important caveat to this approach is that we cannot assume that elongation is purely random assembly. This is because stacking effects will favor the elongation of some sequences in preference to others and the nonrandom trajectory of self-assembly may also be sensitive to initial conditions. Template elongation can also be expected to generate nonrandom motif distributions. The measurements listed in Table 1 were made as follows. All possible motifs of a given length were enumerated. The frequency of each motif and its nearest neighbors (at Hamming distance 1; in the reactor at the end of each run) was counted and stored. The motif defining the largest of the clusters was then determined, and its frequency listed in Table 1 as a percentage of the total frequency of motifs in all clusters. This was then compared against a random model in which the final sequence state of the reactor was constructed as follows. Nucleotide pairs were chosen at random and exchanged between templates, a total of 20,000 exchanges being made. One hundred such random models were generated, and the figure on the right in each entry in Table 1 shows the mean and standard deviation of the largest motif cluster obtained from the random model. Entries in boldface show where the experimental model contains a largest motif cluster that is >3 standard deviations from the mean expected from a random model.
In the nontidal case at high monomer concentrations, for all oligomers up to 5-mers, the most common motif cluster was always present at a frequency at least 3 standard deviations greater than the mean expected from the random models. This suggests that some nonrandom sequence elongation or replication is likely to have been occurring. However, no significant clustering was observed in the nontidal model at lower monomer concentrations. Tides at high monomer concentrations resulted in a loss of significant clustering, whereas tidal effects at low monomer concentrations increased the extent of clustering compared to the nontidal, low monomer concentration case.
Conclusion
In conclusion, replication of oligomers is unreliable compared with de novo sequence generation; see Fig. 15. This is due to a combinatorial explosion of elongation events that occurs with increasing template length. Denaturation-renaturation cycles, irrespective of their cause, promote elongation at splint junctions at both high and low monomer concentrations. At high monomer concentrations, tidal cycling decreases the extent of clustering in motif space, whereas at low monomer concentrations it has the opposite effect, i.e., to increase clustering in motif space. This may be due to either replication effects or nonrandom self-assembly effects, but other methods are required to explore the reaction mechanisms responsible for such clustering. For example, it may be possible in a software model to directly tag sequence segments that have acted as templates, to measure the number of replication cycles each segment takes part in before becoming sequestered. Such techniques are not possible in real chemistry. The same elongation problem arises with temperature oscillation and with monomer concentration oscillation. Experiments have been conducted with a wide range of parameter settings, and the finding that elongation outcompetes replication is extremely robust.

Three OR-coupled (Gánti 2003) autocatalytic cycles potentially capable of mediating template replication are shown by solid arrows. Side-reactions are partially due to p-bond breakage (thin-dashed arrows) but are primarily due to elongation. Only a sample of the possible elongation reactions is shown (thick-dashed arrows). The leftmost cycle must turn four times to supply the matter for one turn of the rightmost cycle.
The stochastic model is limited in some important ways. It considers only simple secondary structures. It cannot be guaranteed that the h-bond dynamics have been run to a complete steady state between p-bond events, however, test runs with longer times allowed for h-bond dynamics to reach steady state revealed no differences in outcome. Rare p-bond forming configurations that have a high p-bond forming propensity will be underrepresented in the microstate sampling, but this bias is expected to vanish in the limit of high sample size.
The model demonstrates that tidal cycling cannot work without acknowledging the need for more complex RNA secondary structures, e.g., hairpins that may be capable of altering the rate of flux between the elongation and the replication pathways. This is because of the ubiquitous presence of elongating side-reactions. One obvious means by which flux could be channeled from the elongation to the replication pathway would be if certain sequences were able to cut themselves out of elongating templates, i.e., by acting as restriction ribozymes.Footnote 6 We hypothesize that a self-splicing replicase ribozyme in an elongation-favoring environment can be shorter and therefore easier to evolve than a replicase ribozyme with ligase activity (Jeffries et al. 1989). Whatever the precise mechanism of the minimal ribozyme, and whenever reaction conditions were such that nonenzymatic ligation could take place, there would have been strong selective pressure for a minimal replicase ribozyme to avoid entrapment in elongating sequences. We have demonstrated that even if these conditions involved tidal cycling or another kind of denaturation-renaturation cycle, the mechanisms of replication hitherto proposed (see Fig. 1) are foiled by the elongation problem. One proposed solution to the elongation problem is the early evolution of a minimal replicase ribozyme with restriction activity.
Notes
Avogadro’s number × Volume is defined as 10,000. Dividing the number of molecules by this value gives the concentration as moles per liter.
The rules are applied to the system using a variant of the SSA algorithm (Elf and Ehrenburg 2004), based on the next reaction method of the Gillespie algorithm (Gillespie 1977; Gibson and Bruck 2000).
These are codes for the classes of equivalent h-bond neighborhood states that contribute the same stacking stability to the central h-bond. They label the 16 configurations shown in Fig. 4 rule 1.
We model a replicase ribozyme that behaves similarly to the sequence-nonspecific RNA-dependent RNA polymerase protein enzyme from Q beta.
Elongation is observed even in the absence of spontaneous ligation if the system is initialized with 10-mers.
Sequence-independent degradation reactions are insufficient because they would result in stochastic loss of sequence information.
References
Braun D, Libchaber A (2004) Thermal force approach to molecular evolution. Phys Biol 1:1–8
Breivik J (2001) Self-organization of template-replicating polymers and the spontaneous rise of genetic information. Entropy 3:273–279
Cantor CR, Schimmel PR (1980) Statistical mechanics and kinetics of nucleic acid interactions. In: Biophysical chemistry. W. H Freeman, San Fransisco, pp 1183–1264
Cech TR (2002) Ribozyme, the first 20 years. Biochem Soc Trans 30:1162–1166
Cruz P, Bubienko E, Borer P (1982) A model for base overlap in RNA. Nature 298:198–200
Elf J, Ehrenberg M (2004) Spontaneous seperation of bi-stable biochemical systems into spatial domains of opposite phases. IEE Syst Biol 1(2):230–236
Ferris JP, Hill AR Jr, Liu R, Orgel LE (1996) Synthesis of long prebiotic oligomers on mineral surfaces. Nature 381:59–61
Fernando CT, Di Paolo E (2004) A model for the origin of long RNA templates. In: Proceedings of the Ninth International Conference of Artificial Life, Boston, MA, pp 1–9
Gánti T (1979) A theory of biochemical supersystems and its application to problems of natural and artifical biogenesis. Akadémiai Kiadó, Budapest/University Park Press, Baltimore
Gánti T (2003) The principles of life. Oxford University Press, Oxford
Gibson A, Bruck G (2000) Efficient exact stochastic simulation of chemical systems with many species and many channels. J Phys Chem A 104:1876–1889
Gilbert W (1986) The RNA world. Nature 319:618
Gillespie D (1977) Exact stochastic simulation of coupled chemical reactions. J Phys Chem 8:2340–2381
Griffith S, Goldwater D, Jacobson JM (2005) Robotics: self-replication from random parts. Nature 437:636
Jeffries AC, Symons RH (1989) A catalytic 13-mer ribozyme. Nucleic Acids Res 17:1371–1377
Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP (2001) RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science 292:1319–1325
Kanavarioti A (1994) Template-directed chemistry and the origins of the RNA world. Origins Life Evol Biosph 24:479–495
Kanavarioti A, Bemasconi C (1990) Computer simulation in template-directed oligonucleotide synthesis. J Mol Evol 31:470–477
Kovac L, Nosek J, Tomaska L (2003) An overlooked riddle of life’s origins: energy-dependent nucleic acid unzipping. J Mol Evol 57:S182—S189
Kuhn H (1972) Selbstorganisation molekularer Systeme und die Evolution des genetischen Apparats. Angew Chem 84:838–862
Lathe R (2003) Fast tidal cycling and the origin of life. Icarus 168:18–22
Lathe R (2005) Tidal chain reaction and the origin of replicating biopolymers. Int J Astrobiol 4(1):19–31
Maynard Smith J, Szathmáry E (1995) The major transitions in evolution. Oxford University Press, Oxford
Reynaldo LP, Vologodskii V, Neri BP, Lyamichev VI (2000) The kinetics of oligonucleotide replacements. J Mol Biol 297:511–520
Rohatgi R, Bartel DP, Szostak JK (1996) Nonenzymatic, template-directed ligation of oligoribonucleotides is highly regioselective for the formation of 3′–5′ phosphodiester bonds. J Am Chem Soc 118:3340–3344
SantaLucia J (1998) A unified view of polymer, dumbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA 95:1460–1465
Schoneborn H, Bulle J, Von Kiedrowski G (2001) Kinetic monitoring of self-replicating systems through measurement of flourescence resonance energy transfer. Chembiochem 12:922–927
Sievers D, Von Kiedrowski G (1994) Self-replication of complementary nucleotide-based oligomers. Nature 369:221–224
Sinclair A, Alkema D, Bell RA, Coddington JM, Hughes DW, Neilson T, Romaniuk PJ (1984) Relative stability of guanosine-cytidine diribonucleotide cores: a h-NMR assessment. Biochemistry 23:2656–2662
Stein DL, Anderson PW (1984) A model for the origin of biological catalysis. Proc Natl Acad Sci USA 81(6):1751–1753
Szathmáry E, Maynard Smith J (1997) From replicators to reproducers: the first major transitions leading to life. J Theor Biol 187:555–571
Szathmáry E, Santos M, Fernando C (2005) Evolutionary potential and requirements for minimal protocells. Topics Curr Chem 259:167–211
Turner DH (2000) Conformational changes. In: Bloomfield VA, Crothers DM, Tinoco I Jr (eds) Nucleic acids: structures, properties and functions. University Science Press
Verga P, Rybicki C, Davis KR (2006) Comment on the paper “Fast Tidal Cycling and the Origin of Life” by Richard Lathe. Icarus 180(1):274–276
Von Kiedrowski G (1986) A self-replicating hexadeoxynucleotide. Angew Chem Int Ed Engl 25:932–934
Von Kiedrowski G (1993) Minimal replicator theory i: parabolic versus exponential growth. Bioorg Chem Front 3:113–146
Wattis J, Coveney P (1999) The origin of the RNA world: a kinetic model. J Phys Chem B 103:4231–4250
Wills P, Kauffman S, Stadler B, Stadler P (1998) Selection dynamics in autocatalytic systems: templates replicating through binary ligation. Bull Math Biol 1:1–26
Xia T, SantaLucia J, Kierzek R, Schroeder R, Cox C, Turner D (1998) Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37:14719–14735
Zielinski W, Orgel L (1987a) Autocatalytic synthesis of a tetranucleotide analogue. Nature 327:346–437
Zielinski W, Orgel L (1987b) Oligoaminonucleoside phosphoramidates. Oligomerization of dimers of 3′-amino-3-deoxynucleotides (GC and CG) in aqueous solution. Nucleic Acids Res 15:1699–1715
Zielinski W, Orgel L (1989) The template properties of triphosphraamidates having CG residues. J Mol Evol 29:281–283
Acknowledgments
This work was partly supported by the Hungarian National Research Fund (OTKA T047245), the National Office for Research and Technology (NAP 2005/ KCKHA005) of Hungary, and the ESIGNET European 6th Framework Grant for Cell Signalling Networks. Thanks go to Johan Elf, Mans Ehrenberg, and Simon McGregor for help with the writing of the code for the stochastic algorithm and design of the two-timescale method. Thanks are due to Richard Lathe for helpful comments during the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Niles Lehman
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Fernando, C., Von Kiedrowski, G. & Szathmáry, E. A Stochastic Model of Nonenzymatic Nucleic Acid Replication: “Elongators” Sequester Replicators. J Mol Evol 64, 572–585 (2007). https://doi.org/10.1007/s00239-006-0218-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0218-4