
Abstract
The structure and evolution of the mtDNA control region (CR) and its flanking genes in economically important dipterans from the family Muscidae (Brachycera: Calyptratae), Haematobia irritans, Musca domestica, Atherigona orientalis, and Stomoxys calcitrans are presented in this paper, along with the description of short noncoding intergenic regions possibly related to CR flanking sequences in Stomoxys calcitrans and Ophyra aenescens mtDNAs (ScIR and OaIR, respectively). S. calcitrans showed a large CR with an ∼550-bp element tandemly repeated and a duplicated tRNAIle gene. The characterization of H. irritans, M. domestica, A. orientalis, and S. calcitrans CR sequences led to the identification of seven conserved sequence blocks homologous to the elements previously described for Calliphoridae and Oestridae species (Brachycera: Calyptratae). Comparative analysis with Drosophila species (Brachycera: Acalyptratae) revealed four conserved regions. The putative functional roles of the conserved elements in the regulation of replication and transcription processes are addressed. The characterization of the structural organization of the mitochondrial genome CR demonstrates the plasticity of the mtDNA molecule in family Muscidae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The mitochondrial DNA (mtDNA) of Metazoan species has a circular, compact organization, conserved gene content, predominantly maternal inheritance, a lack of recombination events, and a high rate of nucleotide substitutions. These features have favored its use as a molecular marker in evolutionary, phylogenetic and population genetics studies concerning a wide range of taxa (Wolstenholme 1992; Saccone et al. 1999; Boore 1999). Animal mtDNA usually contains 37 genes (13 protein-coding genes, 2 ribosomal RNA [rRNA] genes and 22 transfer RNA [tRNA] genes) and a major noncoding region (Boore 1999). The major noncoding region of vertebrate mtDNA contains mitochondrial transcription and mtDNA replication origin promoters (Tracy and Stern 1995; Shadel and Clayton 1997; Taanman 1999). Consequently, it is known as the control region (CR). Despite the fact that the major noncoding region of other animal mtDNA lacks any similarity to vertebrate regulatory elements, they are generally known as the CR as well (Boore 1999).
The finding that the origin of replication of Drosophila mtDNA maps within the CR (Goddard and Wolstenholme 1978, 1980) led to the detection of conserved elements in the CR of insect mtDNA (also known as the A+T-rich region), which may be involved in replication and transcription processes of this genome (Zhang and Hewitt 1997). Recently, Saito et al. (2005) demonstrated the relationship between CR conserved elements and the precise position of the replication origin of seven insect mtDNAs.
Zhang and Hewitt (1997) proposed two insect CR groups: group 1, containing two domains—a conserved region near the tRNAMet, tRNAGln, and tRNAIle (MQI) clustered genes and a variable region adjacent to the 12S rRNA gene; and group 2, with conserved elements distributed along the entire CR sequence. Those structural elements seem to be conserved even in the more basal insect orders (Schultheis et al. 2002).
Aside from genus Drosophila, from which the CR sequences of 12 species have been partially or completely sequenced (Clary and Wolstenholme 1985, 1987; Monnerot et al. 1990; Monforte et al. 1993; Lewis et al. 1994; Inohira et al. 1997; Brehm et al. 2001; Tsujino et al. 2002), only two other dipteran taxa have a fully characterized CR: genus Anopheles (Nematocera), in which the entire CR sequence is very well conserved (Caccone et al. 1996), and Calyptratae flies from families Calliphoridae and Oestridae (Lessinger and Azeredo-Espin 2000; Lessinger et al. 2004), whose CR structure resembles that of Drosophila species (or group 2). Lessinger and Azeredo-Espin (2000) identified eight conserved sequence blocks (CSB) in the CR conserved domain of seven myiasis-causing flies (Calyptratae).
One of the most common events observed in insect mtDNA has been the occurrence of tandem repeats as part of CR sequences. Tandem repeat unit size and copy number variation are responsible for size variation among CR sequences from different species, or even from individuals of the same species (Zhang and Hewitt 1997). Analysis of this in tandem variation among different geographic samples may reveal important data on the population structure of an insect species (Dotson and Beard 2001; Snäll et al. 2002; Mardulyn et al. 2003).
In this paper, we analyze the mtDNA CR and adjacent genes of economically important Calyptratae flies from family Muscidae (superfamily Muscoidea), comparing them with previously described sequences from families Calliphoridae and Oestridae (superfamily Oestroidea) and from Drosophila species (Acalyptratae: Drosophilidae). The taxa Calyptratae and Acalyptratae constitute one of the main sections of suborder Brachycera. The CRs of 11 individuals of the horn fly, Haematobia irritans, 2 of the house fly, Musca domestica, and 2 of the pepper fruit fly, Atherigona orientalis, were entirely sequenced, and the evolution and structural organization of Brachycera mtDNA CR are discussed. In the stable fly, Stomoxys calcitrans, and in the black dump fly, Ophyra aenescens, the comparative analysis of putative CR sequences suggests a different organization of the mtDNA CR and its flanking genes and the presence of an intergenic region. The structural characterization of Muscidae CR will increase our knowledge about animal mitochondrial genome diversity, evolution, and function.
Materials and Methods
Samples and DNA Extraction
Muscidae samples consisted of 11 H. irritans individuals from different geographic localities in South America and Malaysia, 2 M. domestica, 2 A. orientalis, 1 O. aenescens, and 1 S. calcitrans individuals (Table 1). Total individual DNA was extracted from pupae or adults as described by Infante-Vargas and Azeredo-Espin (1995), using a phenol/chloroform procedure, with volumes adapted for microcentrifuge tubes.
PCR Amplifications
The primers used in the amplification reactions for CR sequences and flanking regions for the Muscidae mtDNA are listed in Table 1 and indicated in Fig. 1A. PCRs were done using a PTC-200, MiniCycler (MJ Research), or GeneAmp PCR System 9600 (Perkin Elmer) thermocycler, under the following conditions: 1 μl of template was used in a 25-μl reaction with 1.25 U of Taq DNA polymerase (Invitrogen), a 200 μM concentration of each dNTP, 1.5 mM MgCl2, and a 500 nM concentration of each primer. After an initial denaturation step at 95°C for 5 min, 35 cycles were run, each consisting of melting at 95°C for 1 min, annealing at 45o–52°C for 1 min, and extension at 60°C for 2 min. A final extension step was run at 60°C for 5 min. These conditions are described by Oliveira et al. (2006). Except for S. calcitrans reactions, PCR templates were an aliquot of total individual DNA. The use of total individual DNA of S. calcitrans samples has provided no amplification products. To improve the amplification of S. calcitrans sequences, the DNA template was a 1:500 dilution from the Long-PCR amplification product (∼9.5 kb) of S. calcitrans mtDNA (Barau et al. 2005).

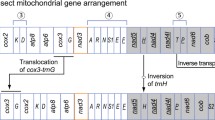
A Schematic organization of insect mtDNA CR and adjacent genes, indicating the primers used for standard PCR in this study (see Table 1 for primer combinations). The primer nomenclature follows Simon et al. (1994). *Primers described by Oliveira et al. (2006); # primers described by Simon et al. (1994). ND2, M, Q, I ,and 12S rRNA represent genes for subunit 2 of NADH dehydrogenase, tRNAMet, tRNAGln, tRNAIle, and subunit 12S of rRNA, respectively. B Structural organization of the mitochondrial genome of the five Muscidae species studied here recovered by standard PCR. The conserved structural elements reported in the control region (CR) sequences of H. irritans, M. domestica, and A. orientalis homologous to the conserved sequence blocks (CSB) of myiasis-causing flies are shown in black circles and numbered 1, 2, and 4–8. S and G elements represent the positions of the predicted secondary structures and of the G islands, respectively. TRU, tandem repeat unit. The bold lines above the CR sequences indicate brachyceran conserved regions (BCRs) I–IV. H. irritans Types 1–3 and A. orientalis Types 1 and 2 refer to the intraspecific CR size variations. OaIR and ScIR represent intergenic noncoding regions in the O. aenescens and S. calcitrans mtDNAs, respectively. C Structural organization of the S. calcitrans mitochondrial genome recovered by assembling mtDNA sequences from a shotgun library. The ScIR represented here is exactly the same region as in B. tRNAIle copies are indicated (I and I2). A gap in S. calcitrans mtDNA is shown as the hatched box (GAP).
Long-PCR Amplification, Restriction Endonucleases Analyses, and mtDNA Shotgun Library
The whole mitochondrial genomes of H. irritans, S. calcitrans, and O. aenescens were amplified via Long-PCR as described by Barau et al. (2005). mtDNA sizes were evidenced by electrophoresis in 0.8% agarosis gel. For S. calcitrans, an aliquot of the Long-PCR products was digested with four restriction endonucleases (HindIII, DdeI, ClaI, and EcoRV), in different reactions, for 4 h at 37°C. The digested fragments were separated by electrophoresis in 2% agarose gel and compared with the restriction profile predicted from S. calcitrans mtDNA sequences (unpublished data from the ongoing mitochondrial genome sequencing project).
As part of a sequencing project initiative, approximately 13 μg of Long-PCR products amplified from the S. calcitrans mtDNA was used to construct a shotgun genomic library, according to the procedure described by Anderson et al. (1982). Long-PCR products were shared by sonication and fragments of 1–2 kb were isolated from a low-melting point agarosis electrophoresis gel. The selected fragments were cloned and sequenced as described below. CR sequences identified by the mitochondrial genome sequencing project were made available to this study.
Cloning and Sequencing
PCR products were purified using 0.05-μm filters, as described by Lessinger and Azeredo-Espin (2000). An aliquot of the purified PCR product was cloned using the TA Cloning kit (Invitrogen), according to the manufacturer’s specifications. The clones were sequenced by automated DNA sequencing in an ABI 377 DNA sequencer using the ABI-PRISM BigDye terminator sequencing v3.1 (Applied Biosystems). Eight sequencing primers were designed based on H. irritans and M. domestica CR sequences: HCR-A (5′ GTAGATAATTTTTTTTTGCG 3′), HCR-A2 (5′ ACTGATAAT TTTCCATTA 3′), HCR-B (5′ TAAAAATTGGTACTATCTCC 3′), HCR-B2 (5′ AAAACAAGCTATTTCTAT3′), MCR-A (5′ TTCTCTATATAAAATTAC 3′), MCR-A2 (5′ AATGTTATTAT TTTTTTC 3′), MCR-B (5′ ATTTATAAAACATACCCC 3′), and MCR-B2 (5′ AAAAAAAAAATTTCGAGA 3′). Three to five clones were sequenced for each amplified region of the Muscidae species.
The 1- to 2-kb purified fragments of S. calcitrans mtDNA were blunted and cloned using the pMOSBlue blunt-ended cloning kit (Amersham Biosciences), according to the manufacturer’s instructions. The clones were sequenced automatically in ABI Prism 377 and 3700 sequencers using the Big Dye terminator kit.
Comparative and Structural Analysis
The nucleotide sequences were submitted to similarity searches in GenBank using BLASTN (Altschul et al. 1997). The tRNA gene sequences were identified using tRNAscan-SE software (Lowe and Eddy 1997). Consensus sequences of the mitochondrial genome of S. calcitrans were assembled using the phred/phrap/consed package (Gordon et al. 1998). The noncoding sequences identified between tRNAIle and 12S rRNA genes were analyzed by searching for structural elements and conserved regions already described for insects (Lewis et al. 1994; Zhang and Hewitt 1997; Inohira et al. 1997; Lessinger and Azeredo-Espin 2000; Brehm et al. 2001; Tsujino et al. 2002), using the software packages Clustal X (Thompson et al. 1997), MEME-motif discovery tool (Bailey and Elkam 1994), and Mfold (Zucker 2003) set to default parameters, with manual adjustments when appropriate. The software Dotlet (Junier and Pagni 2000) was used to identify tandem repeats in CR sequences. Other comparative and statistical data were obtained using MEGA3 software (Kumar et al. 2004).
The nucleotide sequences reported in this paper have the following GenBank accession numbers: AY929377–AY929384 and DQ377072–DQ377074 for H. irritans samples, DQ377075 and DQ377076 for M. domestica, DQ377077 and DQ377078 for A. orientalis, DQ377079 for O. aenescens, and DQ377080 and DQ533708 for S. calcitrans.
Results and Discussion
Structural Conservation of the Muscidae Control Region
Figures 1B and C show schematically the organization of the Muscidae CR sequences. The sizes of O. aenescens and S. calcitrans amplicons recovered by standard PCR were substantially shorter than the CR amplicons of the other species. However, Long-PCR analyses indicated mitochondrial genomes of ∼16 and 18 kb, respectively (see forthcoming discussion). The analysis of CR data retrieved from the S. calcitrans mtDNA sequencing project provides the characterization of almost 60%–70% of the predicted 2.5- to 3-kb CR (938 bp from the 5′ end, flanked by the tRNAMet, tRNAGln and tRNAIle gene cluster, and 839 bp from the 3′ end, flanked by the genes tRNAIle2 [second copy] and 12S rRNA). Searches for CR sequence similarities among H. irritans, M. domestica, A. orientalis, S. calcitrans, and the blowfly species Cochliomyia hominivorax, Co. macellaria, Chrysomya megacephala, Ch. chloropyga, Ch. albiceps, Lucilia eximia (Calliphoridae), and Dermatobia hominis (Oestridae) allowed the identification of seven conserved sequence blocks (CSBs) of eight conserved elements previously described in Calyptratae flies (Lessinger and Azeredo-Espin 2000; Lessinger et al. 2004). CSB 3 was not present in these Muscidae species, possibly representing an Oestroidea-specific motif. Interestingly, CSB 7 was not identified in A. orientalis.
The structural organization of the Muscidae CR resembles that of blowflies, with two domains presenting distinct rates of nucleotide substitutions and indel events: (1) a conserved domain, containing sequence motifs homologous to blowfly CSBs; and (2) a variable domain lacking homology with any other mtDNA sequence. However, we report, in the variable domain of the Muscidae CR, conserved stretches of four or five Gs, previously described by Brehm et al. (2001) in Drosophila species. These G stretches (or G islands) also occur in the blowfly CR variable domain, but they vary in number among species. The Muscidae CR most commonly presents three G islands.
A poly (T) stretch near the MQI genes, TA(A) tandem repeats, and a stem-and-loop secondary structure were insect CR conserved features (Zhang and Hewitt 1997) also identified in Muscidae. The poly(T) stretch (∼20 Ts) corresponds to CSB 1, and the TA(A) region is homologous to CSB 6. The predicted Muscidae secondary structures (Figs. 1B and C; S element) are represented in detail in Fig. 2. In Muscidae species, the most energetically stable predicted structures do not correspond to any CSB, in contrast to the predicted structures described for blowfly CR (Lessinger et al. 2004). Moreover, H. irritans, M. domestica, A. orientalis, and S. calcitrans CR secondary structures are not conserved in primary sequence or relative position (Figs. 1B and C). Nevertheless, these elements are structurally very similar to those of other dipterans (Fig. 2). The involvement of secondary structure in the replication initiation of insect mtDNA was experimentally supported in Orthoptera species (Saito et al. 2005).

The predicted secondary structures identified in the control region (CR) sequences of H. irritans, M. domestica, A. orientalis, and S. calcitrans. The relative positions of these structures are shown as an S in Fig. 1. The free energy values (kcal·mol−1) are shown below each structure.
In order to extend our analysis to other non-Calyptratae dipterans, we compared the mtDNA CR of Muscidae, Calliphoridae, and Oestridae species to those of seven Drosophila species: D. melanogaster (GenBank accession number J01404), D. yakuba (NC_001322), D. subobscura (AJ132900), D. madeirensis (AJ132902), D. guanche (AJ132901), D. teissieri (X54011), and D. virilis (X05914). In addition to the previously described G island motifs (Brehm et al. 2001), four other conserved regions were identified. These regions were named “brachyceran conserved regions” (BCRs), due to their potential to be recognizable in other brachyceran CRs. BCRs I and II are extensive arrays of Ts located on opposite DNA strands, which correspond to CSB 1 and 8, respectively (Figs. 1B and C). BCRs III and IV seem to be homologous to a 300-bp conserved region that was identified in several Drosophila species with short CRs (Clary and Wolstenholme 1987; Monnerot et al. 1990; Monforte et al. 1993; Brehm et al. 2001) and in the type II repeat element of Drosophila species with longer CRs (Lewis et al. 1994; Inohira et al. 1997; Tsujino et al. 2002). Figure 3 shows the alignment and the consensus sequence of BCRs III and IV.

Nucleotide sequence alignment of brachyceran conserved regions III (A) and IV (B) for the Muscidae, Calliphoridae, Oestridae, and Drosophilidade species studied. Consensus sequences are also shown. A dot in the sequence indicates a nucleotide that is the same as that in the consensus sequence. Dashes represent gaps, and letters represent substitutions. The following symbols were used to indicate degenerated sites in the consensus sequences: R = A/G, W = A/T, M = A/C, K = T/G, Y = T/C, and H = A/C/T.
The identification of conserved sequences among Muscidae, Calliphoridae, Oestridae, and Drosophilidae could indicate the similar functional roles of these elements in regulating higher Diptera mtDNA transcription and replication processes. Clary and Wolstenholme (1987) argued that the location of the initial poly(T) (BCR I) near the MQI genes might promote the recognition of a transcription origin oriented to the flanking tRNA cluster. Similarly, BCR II might regulate transcription through the 12S rRNA direction. Recently, Saito et al. (2005) precisely mapped the replication origin of both strands of Drosophila mtDNA to the first nucleotides downstream of both poly(T) elements (BCR I and II). Other evidence from cross-linking protection experiments (Potter et al. 1980, Pardue et al. 1984), S1 nuclease assays (Monforte et al. 1993; Tsujino et al. 2002), and electron microscopy (Goddard and Wolstenholme 1978, 1980) in Drosophila species mapped the mtDNA replication origin of Drosophila CR in the 300-bp conserved region. We suggest that BCRs III and IV (equivalent to CSBs 2, 5, and 6 of Calyptratae species) are homologous to the Drosophila 300-bp conserved region, reinforcing the suggestion of Lewis et al. (1994) on the site-specific protein-DNA interaction properties in this region. It was remarkable that the predicted secondary structures were consistently located within or near BCRs III and IV in all brachyceran species (Figs. 1B and C) (Clary and Wolstenholme 1987; Monforte et al. 1993; Inohira et al. 1997; Tsujino et al. 2002; Lessinger et al. 2004).
Intraspecific Variability of Control Region Sequences
The analysis of intraspecific variability based on mtDNA CR sequences identified few nucleotide substitutions among South American samples from the same species: uncorrected p-distances ranging from 0% to 0.32% for H. irritans, 0.16% for M. domestica, and 0% for A. orientalis. In contrast, significant sequence divergences were found for H. irritans between South American and Malaysian CR haplotypes (from 9.68% to 9.93%).
Significant CR length variation was evident in H. irritans, and was mainly due to duplicated elements. Most individuals analyzed presented a Type 1 CR (Fig. 1B), with ∼1256 bp. Amplification reactions of large numbers of South America samples of H. irritans (Oliveira et al. 2006) identified the Type 1 CR as the most frequent haplotype. The Type 2 CR was specific to the Malaysian mtDNA, with a CR larger than Type 1 (1315 bp), due to multiple insertions in the variable domain. The H. irritans Type 3 CR was shared by three geographically distant South American samples: Barquisimeto (Venezuela), Libertad (Uruguay), and Caraguatatuba (Brazil) (Table 1), despite its low frequency in these populations. The largest-sized Type 3 CR (∼1704 bp) was the result of an ∼450-bp tandem duplication which has taken place in the variable domain of H. irritans CR. Tandem duplications are a common feature of insect CR (Zhang and Hewitt 1997). Variation in tandem repeat numbers was observed in the CR sequences of the two individuals of A. orientalis (110-bp unit), Types 1 and 2 (Fig. 1B).
The low variability among South American H. irritans CRs and their significant divergence in comparison to the Malaysian type are expected considering the extreme geographic distance between these samples. Historically, H. irritans is a new invader species in the South American continent (Valério and Guimarães 1983), and founder effects could be responsible for the low levels of genetic variation found in these samples. Despite that, the occurrence of multiple introductions could not be discarded; in this case, other factors may be responsible for reduced horn fly variability.
Muscidae Mitochondrial Genome Organization
Extreme size variations among Muscidae species and among individuals from the same species (Table 1 and Figs. 1B and C) were found. The species H. irritans, M. domestica, and A. orientalis presented a structural organization and gene order of the CR and flanking genes which were the same as described for the mitochondrial genome of D. yakuba (Clary and Wolstenholme 1985), considered to be ancestral for arthropod mtDNA. In these genomes, the CR is flanked by tRNA gene cluster MQI and the 12S rRNA gene. Figure 1B illustrates the mtDNA CR sequences and flanking genes of the Muscidae species.
For O. aenescens and S. calcitrans mtDNA, standard PCR procedures consistently amplified extremely short CR amplicons, which could be the result of rearrangements involving CR flanking sequences —and, consequently, primer hybridization sites—or PCR artifacts. Based on this PCR strategy, the gene order found in O. aenescens and S. calcitrans CR flanking genes was the same as described for other dipterans (Fig. 1B), but noncoding regions of 382 and 96 bp were identified between MQI and 12S genes in the mtDNA of O. aenescens and S. calcitrans, respectively. These noncoding regions could not be assigned as the putative mtDNA CR, as they lack the conserved elements described for insect CRs in general (Zhang and Hewitt 1997), for Calyptratae dipterans (Lessinger and Azeredo-Espin 2000; Lessinger et al. 2004), and for Muscidae species (discussed above). Searches for sequence similarities in GenBank provided no identity match regarding these noncoding regions.
Different results arise from Long-PCR approaches: the O. aenescens mitochondrial genome amplified by Long-PCR was similar in size to the mtDNA of other Muscidae species (∼16 kb), which diverge from the size expected for a mtDNA molecule with a short CR. Even more interestingly, Long-PCR amplifications of S. calcitrans samples revealed a mtDNA molecule 1.5–2 kb larger than other Muscidae mitochondrial genomes. Restriction endonuclease mapping of S. calcitrans mtDNA Long-PCR amplicons have already suggested that the extra 1.5- to 2-kb sequence was inserted between the ND2 and the 12S rRNA genes (data not shown). This finding was confirmed by the analysis of CR sequences retrieved from the S. calcitrans mitochondrial genome sequencing project, in addition to the description of new features, such as tandem repeats of about 550 bp containing the BCR III and IV elements (Fig. 1C). The exact number of the repeats is still unknown, but they may account for the difference in size of S. calcitrans (∼18 kb) and other dipteran mitochondrial genomes.
Another unusual feature found in the mtDNA of S. calcitrans is the presence of two copies of the tRNAIle gene, one (tRNAIle) as part of the MQI gene cluster—a more conservative position regarding insect mtDNA gene order—and another one flanking the CR 3′ end, near the 12S rRNA gene (tRNAIle2). Both copies are identical, and a possible result from the tandem duplication-random loss model proposed by Macey et al. (1998). Such an event would have required the duplication of the entire CR along with the tRNAIle gene, with subsequent deletions of duplicated CR sequences and conservation of tRNAIle copies. A similar structure was described for blowflies of the genus Chrysomya (Lessinger et al. 2004), illustrating the fact that this phenomenon is not restricted to a single taxon and may reflect a hotspot of structural evolution.
Between the tRNAIle2 and the 12S rRNA genes, we identified the same 96-bp noncoding region (ScIR) that was previously characterized by sequencing standard PCR products (Fig. 1B). However, the gene arrangement described in Fig. 1B for S. calcitrans mtDNA was not recovered by the results retrieved from the mtDNA sequencing project. Technical and methodological difficulties in accessing CR sequence information interfere with both strategies applied for accessing S. calcitrans CR, but we believe that the CR data from the shotgun library analysis provided more reliable results.
Conclusions
Although the PCR strategies used to recover mtDNA CR sequences of the Muscidae species H. irritans, M. domestica, and A. orientalis were successful, structural features related to O. aenescens and S. calcitrans mtDNA limited access to CR data. The Long-PCR amplifications of the complete mitochondrial genomes of O. aenescens and S. calcitrans recovered genomes of ∼16 and 18 kb, suggesting that their CR or flanking genes (ND2, M, Q, I, and 12S) may be involved in duplications and/or rearrangements that occurred during the evolution of the mtDNA structural organization. For S. calcitrans, duplications of CR internal elements and the tRNAIle gene, and the presence of ScIR were confirmed by the analysis of partial results retrieved from an ongoing mitochondrial genome sequencing project. However, the 0.8- to 1.5-kb region that is still missing (GAP; Fig. 1C) in S. calcitrans mtDNA can contain much information about the structural organization of this genome, and we cannot discard the structure reported in Figure 1B by addressing it as a PCR artifact. Despite the presentation of valuable CR data for the stable fly, a final picture of its structure and evolution will depend on the success of whole-mitochondrial genome sequencing projects and technical improvements in PCR and cloning strategies for highly repetitive genomic regions.
Although the duplicated tRNAIle genes of S. calcitrans are conserved and may represent functional copies, it is still unclear whether gene conversion or selective constraints operate to maintain conserved tRNAIle gene copies in the S. calcitrans mitochondrial genome. These results imply a structural plasticity and diversity in the mtDNA molecule of the family Muscidae, and should contribute to further understanding of insect mitochondrial genome evolution.
The recognition of previously described CSBs of myiasis-causing flies (Brachycera: Calyptratae: Calliphoridae, and Oestridae) in the mtDNA CR of H. irritans, M. domestica, A. orientalis, and S. calcitrans (Brachycera: Calyptratae: Muscidae), in addition to the identification of conserved regions shared with Drosophila species (Brachycera: Acalyptratae: Drosophilidae), supports the suggestion that these conserved sequences are maintained by selective functional constraints, due to their potential role as cis-regulatory elements that orient mtDNA replication and/or transcription processes. The identification of these structural homologies among Muscidae, Calliphoridae, Oestridae, and Drosophilidae implies that Brachyceran species have a similar mechanism for the regulation of these processes.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein data base search programs. Nucleic Acid Res 25:3389–3402
Anderson S, de Brujin MHL, Coulson AR, Eperon IC, Sanger F, Young IG (1982) Complete sequence of bovine mitochondrial DNA. Conserved features of the mammalian genome. J Mol Evol 156:683–717
Bailey TL, Elkam C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymer. In: Proceedings of the Second International Congress on Intelligent System for Molecular Biology. AAAI Press, Menlo Park, CA, pp 28–36
Barau JG, Azeredo-Espin AML, Lessinger AC (2005) Conservation and versatility of a new set of primers for long-PCR amplification of complete insect mitochondrial genomes based on Haematobia irritans mtDNA sequences. Mol Ecol Notes 5:885–887
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Brehm A, Harris DJ, Hernández M, Cabrera VM, Larruga JM, Pinto FM, González AM (2001) Structure and evolution of the mitochondrial DNA complete control region in the Drosophila subobscura subgroup. Insect Mol Biol 10:573–578
Caccone A, García BA, Powell JR (1996) Evolution of mitochondrial DNA control region in the Anopheles gambie complex. Insect Mol Biol 5:51–59
Clary DO, Wolstenholme DR (1985) The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization and genetic code. J Mol Evol 22:252–271
Clary DO, Wolstenholme DR (1987) Drosophila mitochondrial DNA: conserved sequences in the A+T–rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J Mol Evol 25:116–125
Dotson EM, Beard CB (2001) Sequence and organisation of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol 10:205–215
Goddard JM, Wolstenholme DR (1978) Origin and direction of replication in mitochondrial DNA molecules from Drosophila melanogaster. Proc Natl Acad Sci USA 75:3886–3890
Goddard JM, Wolstenholme DR (1980) Origin and direction of replication in the mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res 8:741–757
Gordon D, Abajian C, Green P (1998) CONSED: a graphical tool for sequence finishing. Genome Res 8:195–202
Infante-Vargas ME, Azeredo-Espin AML (1995) Genetic variability in mitochondrial DNA of screwworm, Cochliomyia hominivorax (Diptera: Calliphoridae), from Brazil. Biochem Genet 33:237–256
Inohira K, Hara T, Matsuura ET (1997) Nucleotide sequence divergence in the A+T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana. Mol Biol Evol 14:814–822
Junier T, Pagni M (2000) Dotlet: diagonal plots in a Web browser. Bioinfo Appl Note 16:178–179
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinfo 5:150–163
Lessinger AC, Azeredo-Espin AML (2000) Evolution and structural organization of mitochondrial DNA control region of myiasis-causing flies. Med Vet Entomol 14:71–80
Lessinger AC, Junqueira ACM, Conte FF, Azeredo-Espin AML (2004) Analysis of a conserved duplicated tRNA gene in the mitochondrial genome of blowflies. Gene 339:1–6
Lewis DL, Farr CL, Farquhar AL, Kaguni LS (1994) Sequence, organization, and evolution of the A+T region of Drosophila melanogaster mitochondrial DNA. Mol Biol Evol 11:523–538
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequences. Nucleic Acids Res 25:955–964
Macey JR, Schulte II JA, Larson A, Papenfuss TJ (1998) Tandem duplication via light-strand synthesis may provide a precursor for mitochondrial genomic rearrangement. Mol Biol Evol 15:71–75
Mardulyn P, Termonia A, Milinkovitch MC (2003) Structure and evolution of the mitochondrial control region of leaf beetles (Coleoptera: Chrysomelidae): a hierarchical analysis of nucleotide sequence variation. J Mol Evol 56:38–45
Monforte A, Barrio E, Latorre A (1993) Characterization of the length polymorphism in the A + T-rich region of the Drosophila obscura group species. J Mol Evol 36:214–223
Monnerot M, Solignac M, Wolstenholme DR (1990) Discrepancy in divergence of the mitochondrial and nuclear genomes of Drosophila teissieri and Drosophila yakuba. J Mol Evol 30:500–508
Oliveira MT, da Rosa AC, Azeredo-Espin AML, Lessinger AC (2006) Improving access to the control region and tRNA gene clusters of dipteran mitochondrial DNA. J Med Entomol 43:636–639
Pardue ML, Fostel JM, Cech TR (1984) DNA-protein interactions in the Drosophila virilis mitochondrial chromosome. Nucleic Acids Res 12:1991–1999
Potter DA, Fostel JM, Berninger M, Pardue ML, Cech TR (1980) DNA-protein interactions in the Drosophila melanogaster mitochondrial genome as deduced from trimethylpsoralen crosslinking patterns. Proc Natl Acad Sci USA 77:4118–4122
Saccone C, De Giorgi C, Gissi C, Pesole G, Reyes A (1999) Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene 238:195–209
Saito S, Tamura K, Aotsuka T (2005) Replication origin of mitochondrial DNA in insects. Genetics 171:1695–1705
Schultheis AS, Weigt LA, Hendricks AC (2002) Arrangement and structural conservation of the mitochondrial control region of two species of Plecoptera: utility of tandem repeat-containing regions in studies of population genetics and evolutionary history. Insect Mol Biol 11:605–610
Shadel GS, Clayton DA (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66:409–435
Snäll N, Huoponen K, Savontaus M-L, Ruohomäki K (2002) Tandem repeats and length variation in the mitochondrial DNA control region of Epirrita autumnata (Lepidoptera: Geometridae). Genome 45:855–861
Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tracy RL, Stern DB (1995) Mitochondrial transcription initiation: promoter structures and RNA polymerases. Curr Genet 28:205–216
Tsujino F, Kosemura A, Inohira K, Hara T, Otsuka YF, Obara MK, Matsuura ET (2002) Evolution of the A+T-rich region of mitochondrial DNA in the melanogaster species subgroup of Drosophila. J Mol Evol 55:573–583
Valério JR, Guimarães JH (1983) Sobre a ocorrência de uma nova praga, Haematobia irritans (L.) (Diptera: Muscidae), no Brasil. Revta Bras Zool 1:417–418
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141:173–216
Zhang D-X, Hewitt GM (1997) Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol 25:99–120
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:1–10
Acknowledgments
The authors thank those who provided Muscidae samples: Mariana L. Lyra, Pablo Fresia, Santos Gama, Stella Lanzzeri, Estela Martines, Arnaldo Maggi, Alberto Guglielmone, Alfredo Coronado, Patrícia Thyssen, and Ana C. M. Junqueira. We also thank Rosângela A. Rodrigues, Cristina F. Abreu, Joan G. Barau, Pedro C. Feijão, and Fabio O. Dias for technical assistance and Cláudio J. B. Carvalho and Arício X. Linhares for the taxonomic identification of O. aenescens and A. orientalis, respectively. This work, as well as A.C.L., was supported by the Programa Especial de Estímulo à Fixação de Doutores sponsored by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (PROFIX/CNPq; Grant 540.602/01-9). A.M.L.A.E. was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; Grant 03/01458-9), and M.T.O. by a fellowship from Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Martin Kreitman]
Rights and permissions
About this article
Cite this article
Oliveira, M.T., Azeredo-Espin, A.M.L. & Lessinger, A.C. The Mitochondrial DNA Control Region of Muscidae Flies: Evolution and Structural Conservation in a Dipteran Context. J Mol Evol 64, 519–527 (2007). https://doi.org/10.1007/s00239-006-0099-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0099-6