
Abstract
The Mannheimia subclades belong to the same bacterial genus but have taken divergent paths toward their distinct lifestyles. M. haemolytica + M. glucosida are potential pathogens of the respiratory tract in the mammalian suborder Ruminantia, whereas M. ruminalis, the supposed sister group, lives as a commensal in the ovine rumen. We have tested the hypothesis that horizontal gene transfer of the leukotoxin operon has catalyzed pathogenic adaptation and speciation of M. haemolytica + M. glucosida, or other major subclades, by using a strategy that combines compositional and phylogenetic methods. We show that it has been vertically inherited from the last common ancestor of the diverging Mannheimia subclades, although several strains belonging to M. ruminalis have lost the operon. Our analyses support that divergence within M. ruminalis following colonization of the ovine rumen was very rapid and that functional decay of most of the leukotoxin operons occurred early when the adaptation to the rumen was fastest, suggesting that antagonistic pleiotropy was the main contributor to losses in the radiating lineages of M. ruminalis. To sum up, the scenario derived from these analyses reflects two aspects. On one hand, it opposes the hypothesis of horizontal gene transfer as a catalyst of pathogenic adaptation and speciation. On the other hand, it indicates that losses of the leukotoxin operons in the radiating lineages of M. ruminalis have catalyzed their adaptation to a commensal environment and reproductive isolation (speciation).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The leukotoxin (LktA) protein of Mannheimia haemolytica belongs to the E. coli HlyA-like subfamily of cytotoxic RTX (repeats in toxin) proteins present in a range of Gram-negative bacteria (Lo et al. 1987). These RTX proteins all undergo posttranslational fatty acylation and conformational changes as part of maturing their biological behavior (Welch 2001). The subfamily is characterized by a conserved RTX domain (GGXGXDX(L/F/I)X; single-letter amino acid code) that is shared by most substrates of the type I secretion system and by a propensity for multiple interactions with receptors which define their functional properties (Welch 2001). The leukotoxin (lkt) operon codes for four proteins: an internal acyltransferase, encoded by lktC (Lo et al. 1987); a structural RTX toxin, encoded by lktA (Lo et al. 1987); an inner membrane protein with a cytoplasmic ATP-binding cassette (ABC) domain, encoded by lktB, which pumps out LktA protein via interaction with the C terminus of LktA (Highlander et al. 1989); and a membrane fusion protein, encoded by lktD, which forms a bridge between the inner and outer membranes (Highlander et al. 1989). The genes for these four proteins are physically adjacent on the chromosome and are transcribed as lktCA or lktCABD messages (Strathdee and Lo 1989; Highlander et al. 1990).
LktA is essential in both evasion and exploitation of the adaptive immune system during pulmonary infection (Petras et al. 1995; Tatum et al. 1998; Highlander et al. 2000). The LktA protein elicits a number of responses in ruminant target cells derived from the pluripotent hematopoietic stem cell, including polymorphnuclear leukocytes (PMNs; neutrophils), lymphocytes, macrophages, and red blood cells (Kaehler et al. 1980; Shewen and Wilkie 1982; Chang et al. 1986). The biological effects against leukocytes result from activation and apoptosis at sublytic doses or membrane disruption and leakage of content at lytic doses (Jeyaseelan et al. 2001; Deshpande et al. 2002; Leite et al. 2002).
Incorporation of foreign DNA is a major force in the evolution of gene content of bacterial species (Koonin et al. 2001; Boucher et al. 2003). These events are generally thought to catalyze adaptive evolution by one of two molecular mechanisms: (i) RecA-dependent homologous recombination between large repeats (≥25 bp) that may be very far apart in the chromosome; or (ii) illegitimate recombination between closely spaced repeats. Under homologous recombination, gene transfer events can increase the frequency of beneficial alleles via selective sweeps leading to increased fitness of the recipient within its current niche (Lawrence 2002). This mechanism is constrained by the evolutionary distance (DNA sequence divergence) between the two mating partners and the cellular machineries involved (Vulic et al. 1997, 1999). Independent of homologous recombination, evolutionary novelty can be accomplished by incorporation of nonhomologous genes or operons via illegitimate recombination in a process known as horizontal gene transfer (HGT), allowing the recipient to exploit niches that are controlled by unpredictable selective processes (Lawrence and Roth 1996; Lawrence 1997, 1999). In the most extreme cases of HGT, the recipient and its maternal parent become reproductively isolated, supporting a role for HGT as a catalyst of speciation (Lawrence 2002).
A number of observations suggests that the lkt operon from genus Mannheimia has been acquired by HGT leading to pathogenic adaptation and speciation. An early study on the distribution of the lkt operon was made by Burrows et al. (1993), who found that the operon was present in the species M. haemolytica and M. glucosida and in the more distantly related taxon [Pasteurella] trehalosi. The M. haemolytica + M. glucosida group forms one of the most recently diverged subclades within genus Mannheimia based on 16S rRNA sequences, and its species are all potential pathogens in the mammalian suborder Ruminantia (Angen et al. 1999). Interestingly, the CD11a/CD18 (αLβ2) subunits of the lymphocyte function-associated antigen-1 (LFA-1) were identified as a Ruminantia-specific receptor for LktA on leukocytes (Lally et al. 1997; Ambagala et al. 1999; Li et al. 1999; Jeyaseelan et al. 2000), supporting that HGT of the lkt operon could have acted as a mechanism of pathogenic adaptation and speciation. The evolution of the lkt operon was revisited by Davies et al. (2002), who used phylogenetic methods to compare 6952 bp of the lkt operon from M. haemolytica, M. glucosida biogroup 3B, and [P.] trehalosi and noted that the ancestral bovine operon from M. haemolytica has been acquired by HGT from a more distantly related donor. The reality of this HGT event should be questioned, however, as the data said to support Mannheimia’s gain of the lkt operon are ambiguous. For example, the fact that M. ruminalis, the supposed sister group of M. haemolytica + M. glucosida, lives as a commensal in the ovine rumen (Angen et al. 1999) supports a HGT event into the last common ancestor of M. haemolytica + M. glucosida (Fig. 1), but the pattern could also be due to altered selection on genes that are involved in evasion and exploitation of the immune system and consequent loss of the lkt operon in the M. ruminalis subclade.

Phylogenetic relationships, habitats, lktA genotypes of the five Mannheimia subclades. Redrawn from Angen et al. (1999).
In this article, we rigorously test the hypothesis that the ancestor of M. haemolytica + M. glucosida gained the lkt operon via HGT by using a strategy that combines compositional and phylogenetic methods. First, we identify bifurcation order of the Mannheimia subclades, allowing the lktA genotype and the corresponding β-hemolytic phenotype to be mapped and compared with transitions in key traits (e.g. pathogenicity and niche utilization). Then we test for the origin of the lkt operons by inferring the relationships of partial lktA sequences with homologous sequences from other Gram-negative bacteria. Finally, we rank genes according to their convergence to the average genome signature of M. haemolytica based on the relative 3:1 dinucleotide bias.
Materials and Methods
Taxa Used
For this study we used 58 strains to represent the diversity within genus Mannheimia based on phenotypic characters (biogroups) and geographic origin (see Table 1 for a description of strains used in this study, along with their accession numbers). However, it is possible that the samples are biased because certain geographic areas and certain organismal groups have been more extensively studied than others and therefore are overrepresented in out data sets (e.g., strains belonging to M. ruminalis have been isolated exclusively from the United Kingdom). Most taxa in our analyses have been represented in previous studies by Angen et al. (1997a– c, 1999) and Blackall et al. (2001), and ribotypes and electrophoretic types are given in those publications.
Analysis of Genes, Pseudogenes, and Remnant DNA Sequences
We analyzed the distribution of the lkt operons in genus Mannheimia using Southern blot. We constructed a probe in the +845/+1302 region of lktA from M. haemolytica strain PHL213 by using the forward primer manpop_UP (5′-CCAAAGCCGTTTCTT CTTACA-3′) in conjunction with the reverse primer manpop_DOWN (5′-TAACGGGCRTCGTAACCATT-3′), which were designed from published sequences. The reaction conditions were 2.5 U Taq polymerase, 16 mM (NH4)2SO4, 67 mM Tris–HCl, 0.01% Tween-20, 2.5 mM Mg2SO4, each primer at 0.5 mM, and each nucleotide at 0.1 mM. The cycling conditions were initial denaturation at 94°C followed by 25 cycles at 94°C for 30 s, 52°C for 30 s, and 72°C for 30 s, finishing with extension at 72°C for 10 min. The PCR product was labeled with digoxigenin-11-dUTP using the Random Primed DNA Labeling Kit according to the manufacturer’s instructions (Roche).
To determine inactivation of the lkt operons, we screened the strains for the β-hemolytic phenotype on sheep blood agar as described previously (Murphy et al. 1995).
To determine DNA loss in the inactivated lkt operons from M. ruminalis, we estimated the length of the region in six nonhemolytic strains using a two-part strategy. First, we searched for conserved sequences in the 5′ and 3′ flanking regions of the lkt operon between the genomes of the β-hemolytic strain HPA113 and the nonhemolytic strain HPA92T. We constructed a probe in the 5′ flanking region (hslU gene) of the lkt operon from the published sequence of strain HPA113 (GenBank: AY425280) by using the forward primer HPA113.1_UP (5′-GCGAAAGATCAA TGGGGTAA-3′) in conjunction with the reverse primer HPA113.1_DOWN (5′-CACGAATCGGTAAACGACCT-3′). The conditions were as for the lktA probe above, but with annealing at 55°C. To construct phagemid libraries for strain HPA92T, we partially digested genomic DNA with the restriction enzyme Sau3AI. Aliquots were run on agarose gels and fragments of approximately 3–6 kb were cloned into the BamHI site of the Zap Express vector according to the manufacturer’s instructions (Stratagene). The ligation mixture was packaged in vitro into the Gigapack III Gold Packaging Extract and transfected into Escherichia coli XL1-Blue MRF′ according to the manufacturer’s instructions (Stratagene). The plaques were lifted and cross-linked to a 0.45-μm nitrocellulose membrane (Millipore) according to the manufacturer’s instructions (Stratagene). The phagemid libraries were screened for hslU clones and the pBK-CMV phagemid vector was excised in vivo from the ZAP Express vector using the ExAssist helper phage and E. coli XLOLR according to the manufacturer’s instructions (Stratagene). The plasmids were then directly sequenced (GenBank AY425275).
To amplify the region in the nonhemolytic M. ruminalis strains, we designed the forward primer rumpop_UP (5′-AATGGTTGA AGCGATGAAGG-3′) and the reverse primer rumpop_DOWN (5′-TTGCGGTAGCCAAGAGAAAG-3′) from conserved sequences in the 5′ and 3′ flanking regions between the genomes of strains HPA113 and HPA92T. We used the Expand Long Template PCR System according to the manufacturer’s instructions (Roche). The cycling conditions were initial denaturation at 94°C followed by 30 cycles at 94°C for 1 min, 52°C for 1 min, and 68°C for 8 min, finishing with extension at 72°C for 10 min. We could not amplify the region from the two nonhemolytic strains HPA81 and HPA93, presumably because substitutions have resulted in imperfect matches of the primers.
Model Fit and Model Selection
The Akaike Information Criterion (AIC) was used to assess how well various substitution and topology models fit the sequence data (Akaike 1973; Burnham and Anderson 2002; Posada and Buckley 2004). The AIC is an estimate of the amount of information that is lost when a given model is used to approximate the full truth (the relative Kullback-Leibler distance). The AIC is a function of the maximized log-likelihood (lnL) and the number of estimated parameters (K) for a model: AIC = –2lnL + 2K, with lower AIC values being better. From AIC, it is also possible to compute Akaike weights (Burnham and Anderson 2002; Posada and Buckley 2004). The Akaike weight of a model can be interpreted as the conditional probability of the model given the data and the set of initial models. It is possible to estimate the relative importance of a model feature by simply summing Akaike weights across the subset of models sharing that feature. Inference can thus be based on a large set of models simultaneously (Burnham and Anderson 2002; Posada and Buckley 2004). Among other things, this is helpful in avoiding the model selection problems associated with misspecification (Zhang 1999). Here, AIC is used to select the best model for construction of phylogenetic trees and for determining whether there is support for a hard polytomy in M. ruminalis.
Reconstruction of Phylogenetic Trees
A species phylogeny was necessary to test the HGT of the lkt operon. The relationships of the strains in this study were inferred with 16S rRNA; these sequences have been used successfully for systematic studies in this group (Angen et al. 1999). Sequences for 19 strains were obtained from GenBank. The primers previously described by Angen et al. (1999) were used to amplify and directly sequence 16S rRNA from 22 additional strains. The 16S rRNA sequences were aligned using Dialign 2 (Morgenstern 1999) with default settings. Phylogenetic trees were reconstructed using Bayesian techniques as implemented in the program MrBayes version 3.1.1 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). The best-fitting model was GTR + invgamma based on the AIC computed by the program MrModeltest version 2.2 (Nylander 2004). Markov chain Monte Carlo (MCMC) was run for 10 million generations with four chains while sampling once every 100 generations. Convergence was confirmed by comparing the results of two independent runs. The program Tracer version 1.3 (Rambaut and Drummond 2004) was used to determine burn-in and also for further confirmation of proper mixing and adequate run-length. A burn-in of 1 million generations (10,000 samples) was used. The distribution of 16S rRNA trees resulting from MCMC was summarized in the form of a consensus tree with all compatible bipartitions included by using the authors’ own software in a manner that is essentially identical to what is obtained when using MrBayes’ sumt command with the setting contype=allcompat, except that branch lengths were averaged over all trees, setting the branch length to zero for those trees that did not contain the corresponding bipartition as suggested by Felsenstein (2003). The tree was rooted based on a maximum likelihood analysis reported by Angen et al. (1999).
To test for the origin of the lkt operons, we inferred the relationships of 101 strains with partial sequences from genes encoding cytotoxic RTX proteins. The +845/+1302 region of lktA from 23 lktA {+} strains was amplified by using the PCR primers manpop_UP and manpop_DOWN. The reaction and cycling conditions were as above. These PCR products were then directly sequenced. We could not amplify lktA from the lktA {+} strain W4672/1, presumably because substitutions have resulted in imperfect matches of the primers. We used the +845/+1302 region of lktA from M. haemolytica strain PHL213 for selecting putative orthologues from the NCBI nonredundant (nr) database by performing TBLASTX searches (Altschul et al. 1990). We identified 78 sequences corresponding to genes encoding cytotoxic RTX proteins (E-value < e−10; score > 99) (see Table 2 for a complete list of retrieved rtxA nucleotide sequences, along with their accession numbers). Phylogenetic trees of the nucleotide sequences were reconstructed in the same way as the 16S rRNA trees. The distribution of rtxA trees from MCMC was summarized in the form of a 50% majority rule consensus tree corresponding to MrBayes’ sumt command with the setting contype=halfcompat. The tree was rooted by using the eight Bordetella sequences as outgroup.
Hard Versus Soft Polytomy
To test for the presence of hard polytomies in M. ruminalis, we explicitly assessed support for a range of alternative tree topologies. There are 11 internal branches in M. ruminalis, 8 of which have posterior probabilities below 95%. Using software written by us, we constructed the 256 possible variants, where one or more of these eight branches have been collapsed. To each of these alternative trees, we then fitted various substitution models using the program baseml from the PAML package version 3.14 (Yang 1997). The models we used were GTR + gamma and four different versions of nparK = 2. The nparK = 2 model accounts for different rates across sites by having several separate rate categories, each applying to a different proportion of sites (Yang and Roberts 1995). Here, we tested versions with two, three, four, or five rate categories. We fitted the different models to each tree topology, in each case recording the number of free parameters (including parameters associated with branch lengths) and the maximized likelihood. To check for convergence and ensure that the global maximum had been found, we fitted each model to each topology in five independent runs. For each model/topology combination, we used the highest of the five independently obtained likelihoods for further computations. This allowed us to calculate AIC and Akaike weights, and from this we could finally compute the cross-model support for each individual tree topology. The main purpose of fitting a range of models was to avoid the errors associated with misspecification (Zhang 1999).
We believe that our strategy holds some advantages over previous methods which were based on classical hypothesis testing (Jackman et al. 1999; Slowinski 2001). The main problem with such approaches is that we are interested in assessing the support for the null hypothesis (polytomy) and this information is not contained in the p value because failure to reject a null model does not necessarily mean that it is well supported. This problem is exacerbated when several internal branches are tested simultaneously and it is problematic how (and whether) to correct for multiple testing artifacts. Thus, conservative correction methods, such as Bonferroni, will result in a bias for the polytomy-containing topologies (Slowinski 2001).
Compositional Analysis
To rank genes according to their convergence to mutational bias in the genome of M. haemolytica, we retrieved nucleotide sequences of genes from the NCBI nonredundant (nr) database. When the source of a gene was not clearly defined, these sequences were excluded from the present study. When multiple alleles of the same gene were retrieved, we used those sequence data as queries to search the preliminary M. haemolytica strain PHL213 genome by using BLASTN (Altschul et al. 1990) with default settings, and only the allele with the highest E-value was included in the present study. In total, we found 56 genes (∼68 kb, corresponding to ∼2.8% of the genome size) using this procedure (see Table 3 for a complete list of retrieved M. haemolytica genes, along with their accession numbers). We used this data set to create an average genome signature (μ) based on the relative 3:1 dinucleotide bias (z) in individual genes as described previously (Hooper and Berg 2002). This method uses a variant of Hotelling’s T 2 statistics as a multivariate distance measure between each gene to the average genome signature and identifies deviant genes at a given significance level.
Sequence Data
Sequences have been deposited in the GenBank database (accession numbers AY425275, AY425282–AY425295, and DQ301920–DQ301950).
Results
3:1 Dinucleotide Bias of the lkt Genes in M. haemolytica
Employing a compositional approach to the identification of horizontally transferred genes requires that genes from the donor and recipient have a different nucleotide composition. Relative abundances in dinucleotide frequencies are unique to most living organisms and consequently constitute a genome signature that is discriminatory between sequences from different organisms (Karlin et al. 1997; Campbell et al. 1999). From these signatures, the distance of an individual gene to the average values of the genome can be calculated. Recent work by Hooper and Berg (2002) redefined this approach by using only the average relative 3:1 dinucleotide abundance, which is the combination that is least affected by codon and amino acid usage. Here we used a data set of 56 genes to create an average genome signature (μ) of M. haemolytica based on the relative 3:1 dinucleotide bias (z) in individual genes. The multivariate distance measure (T 2) between each gene to the average genome signature was calculated (Table 3). The T 2 values were F-distributed with (15,41) degrees of freedom, corresponding to a critical limit of T 2 ∼ 33 (α = 0.1). This means that the method is expected to pick out at least 10% (five or six) of the genes that deviate most from the genomic signature because the significance level is lower in biological data sets (Hooper and Berg 2002). The T 2 values for the lkt genes ranged from 11.22 to 16.07, suggesting that they are nondeviant from the average genome signature of M. haemolytica.
Phylogeny of 16S rRNA Sequences
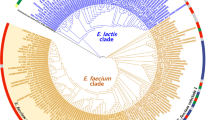
The 16S rRNA data set included 1257 bp after removal of ambiguous bases. Both runs converged and the consensus tree with all compatible bipartitions included and with branch lengths averaged over all trees, setting the branch length to zero for those trees that did not contain the corresponding bipartition, is shown in Fig. 2, along with posterior probabilities (PPs). This phylogram is largely topologically concordant with the maximum likelihood analysis reported by Angen et al. (1999) and indicates moderate to strong PP support for monophyly of the five subclades (M. haemolytica + M. glucosida, M. ruminalis, M. granulomatis, M. varigena, and subclade V comprising unclassified strains) (PP = 82%–100%) and bifurcation order among any subclades (PP = 100%) but very low PP support for bifurcation order among strains within M. ruminalis. There was strong PP support for a sister-group relationship between M. haemolytica + M. glucosida and M. ruminalis as expected (PP = 100%).

Distribution of 16S rRNA trees from MCMC summarized in the form of a consensus tree with all compatible bipartitions included and with branch lengths averaged over all trees, setting the branch length to zero for those trees that did not contain the corresponding bipartition. The tree was rooted based on a maximum likelihood analysis reported by Angen et al. (1999). Posterior probability values are shown for all compatible bipartitions. Presence/absence of the lktA genotype and the corresponding β-hemolytic phenotype is indicated by {+/–;+/–}. Monophyly of the five subclades reported by Angen et al. (1999) is indicated by black circles. Strains present in the lktA tree (Fig. 4) are indicated by asterisks. Nomenclature: sequence names contain abbreviations of the taxonomic group (genus and species) followed by the corresponding strain ID as listed in Table 1.
Hard Versus Soft Polytomy for M. ruminalis
Our Bayesian analyses showed that most internal branches are very short and indicated low PP support for bifurcation order among strains within M. ruminalis (Fig. 2), suggesting simultaneous divergence (hard polytomy) or at least very rapid early divergence (soft polytomy). The hypothesis that the M. ruminalis subclade contains hard polytomies was approached as a model selection problem. First, we constructed the full set of 256 tree topologies where one or more of the eight internal branches with PPs below 95% have been collapsed. Then we used the maximum likelihood principle to fit five different substitution models to each of these alternative tree topologies. Lastly, we calculated the AIC and Akaike weights, and from this we could finally compute the cross-model support for each individual tree topology. The tree topology with the strongest cross-model support (0.225) suggests collapse of seven of the eight internal branches with PPs <95% (Fig. 3). The topology (HPA113 + HPA98) is concordant with the Bayesian analysis (PP = 82%). The next three tree topologies ranked according to their cross-model support (0.081–0.083) all have one less collapsed internal branch (data not shown).

Cladogram showing the M. ruminalis tree topology with the strongest cross-model support. The tree was rooted in the same way as in Fig. 2. Posterior probability values are shown for all compatible bipartitions. Nodes with posterior probabilities above 95% were excluded from the analysis and are indicated by black circles. Nodes with posterior probabilities <95% that were not collapsed by the analysis are indicated by white circles. Hard polytomies are indicated by numbers, suggesting early (1) and late (2) radiation. Presence/absence of the lktA genotype and the corresponding β-hemolytic phenotype is indicated by {+/–;+/–}.
Distribution of the lktA Genotype and Phenotype on the Mannheimia Phylogeny
If the ancestor of M. haemolytica + M. glucosida, or other major subclades, gained the lkt operon via HGT, we would expect that the lktA genotype and the corresponding β-hemolytic phenotype are present only in these subclades. Alternatively, finding that these character states are present in different major subclades would suggest vertical descent, accompanied by major divergences in the lktA genotype and the corresponding β-hemolytic phenotype. The lktA genotype and the corresponding β-hemolytic phenotype are given in Fig. 2 and Table 4. Both character states were present among any subclades, including all strains belonging to M. haemolytica + M. glucosida (15 strains), M. granulomatis (11 strains), and M. varigena (5 strains), but only a fraction of strains belonging to M. ruminalis (7 of 13 strains) and subclade V (13 of 14 strains). All lktA {+} strains were also β-hemolytic, whereas all lktA {−} strains were nonhemolytic.
Determination of DNA Loss in the Inactivated lkt Operons from M. ruminalis
We used a two-part strategy to determine DNA loss in the inactivated lkt operons from M. ruminalis. First, we searched for conserved sequences in the 5′ and 3′ flanking regions of the lkt operon between the genome of the β-hemolytic strain HPA113 and that of the nonhemolytic strain HPA92T and found that the hslU-lapB sequence in the 5′ flanking region and the tauB sequence in the 3′ flanking region of the lkt operon from strain HPA113 were conserved in the sequence hslU-lapB-tauB of strain HPA92T. Then we designed forward and reverse primers in these conserved sequences, allowing us to determine DNA loss in the inactivated lkt operons from nonhemolytic strains (measured as the reduction in amplicon size). The amplicon sizes are given in Table 5, along with their last β-hemolytic ancestor. The amplicon sizes from three of four nonhemolytic strains, including strain HPA92T, were 0.5 kb, corresponding to absence of the entire lkt operon. The amplicon size from the nonhemolytic strain HPA88, along with the β-hemolytic strain HPA113, was 7.9 kb, corresponding to the presence of the entire lkt operon.
Phylogeny of the rtxA Sequences
The distribution of the lkt operon can always be explained by vertical inheritance followed by losses in certain subclades. Phylogenetic reconstructions are therefore necessary to distinguish between this scenario and a scenario whereby the ancestor of M. haemolytica + M. glucosida, or other major subclades, gained the lkt operon via HGT. Loss of the lkt operon is expected to produce phylogenetic trees that broadly resemble the accepted monophyletic organismal groups, whereas HGT is expected to produce trees where the rtxA sequences from the recipient subclades group with homologous sequences from their donors. The rtxA dataset included 458 bp after removal of ambiguous bases. Both runs converged and the 50% majority rule consensus tree is shown in Fig. 4, along with PPs. There was strong support for grouping of the sequences from the Mannheimia and [P.] trehalosi strains (PP = 100%). There was also strong support for grouping most of the sequences from M. haemolytica + M. glucosida and M. ruminalis (except M. glucosida strain P730) and all the sequences from [P.] trehalosi (PP = 100%). Support for bifurcation order of the [P.] trehalosi group and strain BNO311 belonging to subclade V was low (PP = 62%). This means that the [P.] trehalosi sequences form a sister group to most of the sequences from M. haemolytica + M. glucosida and M. ruminalis (except M. glucosida strain P730) and, possibly, strain BNO311.

Distribution of rtxA trees from MCMC summarized in the form of a 50% majority rule consensus tree. The tree was rooted by using the eight Bordetella sequences as outgroup. Posterior probability values are shown for all compatible bipartitions. Grouping of the sequences from the Mannheimia and [P.] trehalosi strains is indicated by a black circle. The sister-group relationship between the [P.] trehalosi sequences and most of the sequences from M. haemolytica + M. glucosida and M. ruminalis (except Mgl_P730) is indicated by a white circle. Grouping of most of the sequences from M. haemolytica + M. glucosida and M. ruminalis (except Mgl_P730), including those sequences belonging to allele group 2, is indicated by a gray circle. Nomenclature: sequence names contain abbreviations of the taxonomic group (genus and species) followed by the corresponding strain ID and/or the accession number as listed in Tables 1 and 2.
Discussion
Vertical Inheritance
Prior to this work, no Mannheimia rtxA sequences from outside M. haemolytica + M. glucosida had been reported and their arrival in this subclade was unclear. Davies et al. (2002) used phylogenetic methods to compare 6952 bp of the lkt operon from M. haemolytica, M. glucosida biogroup 3B, and [P.] trehalosi and noted that the ancestral bovine operon from M. haemolytica has been acquired by HGT from a more distantly related donor. However, such arguments based on a relatively small number of taxa can be misleading because of problems in information content. Our analyses, based on a larger collection of strains and utilizing a combination of compositional and phylogenetic methods, indicate vertical inheritance from the last common ancestor of genus Mannheimia.
Given the collection of strains, information on their lktA genotype and the corresponding β-hemolytic phenotype, and an organismal phylogeny that describes bifurcation order, the characteristics of their ancestors can be inferred. Our analyses revealed that both character states were present among any subclades, including all strains belonging to M. haemolytica + M. glucosida, M. granulomatis, and M. varigena (Fig. 2 and Table 4). These results are compatible with a history of vertical inheritance of the lkt operon from the last common ancestor of genus Mannheimia to any of its descendants followed by losses in the terminal branches of M. ruminalis and subclade V, thus opposing the hypothesis that the ancestor of M. haemolytica + M. glucosida gained the lkt operon via HGT.
Reconstruction of the rtxA tree provided strong support for grouping all of the sequences from Mannheimia, although the analysis also revealed a number of conflicting phylogenetic signals (Fig. 4). This incongruence between gene trees and organismal phylogenies could arise from phylogenetic noise due to convergence and/or sequences with poor phylogenetic signal or from gene transfer events (homologous recombination or HGT). The strong support for a sister-group relationship between the [P.] trehalosi sequences and most of the sequences from M. haemolytica + M. glucosida and M. ruminalis (except M. glucosida strain P730) is interesting because it suggests a history of gene transfer between the ancestor of M. haemolytica + M. glucosida and M. ruminalis and [P.] trehalosi. The lkt operon from the ancestor of these Mannheimia subclades does not seem to have been acquired by HGT from [P.] trehalosi, since this scenario would require loss of the operon prior to the gene transfer event, something that seems unlikely given the presence of the lktA genotype and the corresponding β-hemolytic phenotype among any subclades within genus Mannheimia. Therefore, the incongruence observed in the rtxA tree is compatible only with a history of HGT from the ancestor of M. haemolytica + M. glucosida and M. ruminalis to [P.] trehalosi or homologous replacement of vertically inherited +845/+1302 regions. Previous analyses of the lkt genes from M. haemolytica, M. glucosida biogroup 3B, and [P.] trehalosi have revealed the relevance of gene transfer in shaping the lkt operons (Davies et al. 2001, 2002). Although this work suggested a mosaic origin of the lktBD genes from [P.] trehalosi, the +845/+1302 region of lktA from [P.] trehalosi showed no evidence of recent gene transfer (Davies et al. 2001, 2002). From a phylogenetic perspective, these results concord with the relatively old age of this gene transfer event followed by divergent evolution of the +845/+1302 region of lktA in [P.] trehalosi. In order to determine whether the incongruence observed in the rtxA tree is the result of HGT from the ancestor of M. haemolytica + M. glucosida and M. ruminalis to [P.] trehalosi or homologous replacement of vertically inherited +845/+1302 regions, we need to address the question of the origin of the lkt operon from [P.] trehalosi. However, the unstable position of genus Mannheimia and [P.] trehalosi in the Pasteurellaceae tree (Christensen et al. 2004) and the lack of a compositional signature for [P.] trehalosi limit our ability to evaluate these alternative scenarios.
The results of Davies et al. (2002) also revealed that a 4.4-kb region of the ancestral bovine operon from M. haemolytica strains belonging to the bovine A2 complex, including the entire lktA gene (allele group 2), was more divergent than the homologous sequences from other strains belonging to M. haemolytica, M. glucosida biogroup 3B, and [P.] trehalosi, suggesting that the entire lkt operon has been acquired by HGT from a more distantly related donor. Following Davies and coworkers’ hypothesis, we would expect a robust and systematic incongruence toward the donor. However, we found strong support for grouping most of the rtxA sequences from M. haemolytica + M. glucosida and M. ruminalis (except M. glucosida strain P730), including those sequences belonging to allele group 2 (Fig. 4).
The multivariate distance measure (T 2) between each gene to the average genome signature (μ) of M. haemolytica based on the relative 3:1 dinucleotide bias (z) in individual genes identified the lkt genes as nondeviant. These results also support a history of vertical inheritance of the lkt operon from the last common ancestor of genus Mannheimia to M. haemolytica, thus opposing the hypothesis that the ancestor of M. haemolytica + M. glucosida gained the lkt operon via HGT. However, false negatives (missed transferred genes) arise when genes have ameliorated due to the mutational processes affecting the recipient genome or the genes are closely related to the recipient genome in terms of context bias (Lawrence and Ochman 1997). Therefore, the observed distances between each lkt gene to the average genome signature cannot rule out ancient HGT events, although this is an unlikely scenario, since the data from both phylogenetic methods support a history of vertical inheritance from the last common ancestor of genus Mannheimia.
Radiation in M. ruminalis
In order to explain the discontinuous distribution of the lktA genotype and the corresponding β-hemolytic phenotype in M. ruminalis, it was necessary to resolve the bifurcation order among strains within this subclade. Our expanded analyses allowed us to demonstrate that two hard polytomies are implicated (Fig. 3). The size of the early radiation (measured as the number of new genotypes) is eight, suggesting that divergence following colonization of the ovine rumen was very rapid.
Radiation of a single lineage into a range of genotypes, as appeared to happen once M. ruminalis colonized the rumen, is not surprising given the number of new niches that would be encountered. However, the results presented here do not allow us to test whether radiation reflects the differentiation of a single ancestor into an array of lineages that inhabit a variety of environments and that differ in the morphological and physiological traits used to exploit those environments (adaptive radiation) or rapid proliferation accompanied by negligible or infrequent ecological differentiation (nonadaptive radiation). There is some evidence that strains belonging to M. ruminalis differ in physiological traits (Angen et al. 1997b), but a fit between the diverse phenotypes of descendant lineages and their divergent environments and evidence that those phenotypes are indeed useful where they are employed are needed to fulfill the criteria of adaptive radiation as proposed by Schluter (2000).
Mechanisms of Gene Loss in M. ruminalis
Radiation is consistent with losses of the lkt operons in M. ruminalis. If one assumes that the evolving populations adapted to different ecological niches offering different conditions of growth, then this diversity would imply that different pools of unused gene activities have been lost in those populations.
Genes and gene activities are generally thought to be lost from populations over the course of evolution by one of two evolutionary mechanisms: (i) antagonistic pleiotropy; or (ii) mutation accumulation. Under antagonistic pleiotropy, adaptation to the selective environment and functional decay in other environments are caused by the same mutations. Independent of adaptive mutations, genes can also be lost from a population by the fixation of neutral or nearly neutral mutations via selection-independent genetic drift. This mechanism is thought to account for much of the loss of genes in prokaryotes (Lawrence and Roth 1999; Mira et al. 2001).
These two processes have long been recognized, but their relative contributions are not easy to examine. Our data showed that loss of the β-hemolytic phenotype has occurred in four of eight early radiating lineages (Fig. 3), accounting for 67% of all losses in M. ruminalis. This pattern is consistent with antagonistic pleiotropy, which predicts that most losses should occur early when adaptation to the rumen is fastest and that most lineages should exhibit parallel loss of functions (Cooper and Lenski 2000). We do not expect this association between the dynamics of adaptation and loss via mutation accumulation, which predicts that losses of unused functions should accumulate stochastically at a constant rate that depends only on the mutation rate in the relevant genes (Cooper and Lenski 2000). Our results therefore suggest that inactivation of the lkt operons in the radiating lineages of M. ruminalis have catalyzed their adaptation to a commensal environment and reproductive isolation (speciation), although they do not allow us to experimentally verify that functional decay of the lkt operons is beneficial in the ruminal environment.
The loss of gene content of bacterial species is thought to occur by the same molecular mechanisms that influence their ability to incorporate DNA from other species: (i) RecA-dependent homologous recombination; or (ii) illegitimate recombination. Because the frequency of homologous recombination does not appear to be constrained by the distance between the repeated sequences, it often results in large deletions (Moran and Mira 2001). On the contrary, illegitimate recombination between closely spaced repeats results in multistep deletion of pseudogenes (Andersson and Andersson 2001; Silva et al. 2001). The results of Andersson and Andersson (2001) revealed that the mean and median sizes of the deletions in pseudogenes from four Rickettsia genomes were 51.2 and 4 bp per event, respectively, although two large deletions, of 599 and 767 bp, were observed. However, recent work has shed light on the role of RecA-independent recombination events in fast reductive evolution. Nilsson and coworkers (2005) examined the rate and molecular mechanisms of genome reduction in Salmonella enterica by using serial passage and genetic selections. Those authors showed that the individual chromosomal deletions varied in size from ∼1200 to 202,232 bp. The potential homology at most of the deletion endpoints was ≤12 bp, suggesting that rapid deletion of large block of DNA, including functional genes or operons, could arise from RecA-independent mechanisms.
We identified chromosomal deletions of ∼7400 bp, corresponding to the entire lkt operon, in three of four nonhemolytic strains, including strains HPA98 and HPA109, which diverged only recently from their last β-hemolytic ancestors, whereas we found no evidence for DNA loss in the inactivated lkt operon from strain HPA88 (Fig. 3 and Table 5). The results from strains HPA92T and HPA88, which diverged from the last common ancestor of M. ruminalis, do not allow us to examine DNA loss rates because of problems associated with estimating the age of initial deletion formation. However, following our hypothesis of antagonistic pleiotropy, which predicts that functional decay of most of the lkt operons should occur early when adaptation to the rumen is fastest, it is expected that similar DNA loss rates would result in equal sizes of deletions. Our results therefore support a very low DNA loss rate in strain HPA88, although they do not rule out the alternative scenario of equal DNA loss rates, which implies that inactivation of the lkt operon from strain HPA88 occurred more recently compared to other nonhemolytic strains. This potential discrepancy between the time of divergence from their last β-hemolytic ancestors and the observed sizes of the deletions points toward two most probable explanations. On one hand, it is possible that DNA loss occurs at different rates in the radiating lineages of M. ruminalis due to differences in the molecular mechanisms involved such as those discussed above. On the other hand, the results of Nilsson et al. (2005) showed that the DNA loss rate of WT bacterium was 0.05 bp per chromosome per generation and ∼50-fold higher in a mutS mutant. These data suggest that high DNA loss rates could arise from defects in the methyl-directed mismatch repair (MMR) system. Although there is some evidence that strains belonging to M. ruminalis differ in DNA loss rates, we need to explore the deletion endpoints for repeats and the MMR system for defects in order to estimate the molecular mechanisms responsible for reductive evolution.
Implications for Pathogenic Adaptation and Speciation
The data collected here provide strong support for vertical inheritance of the lkt operon but do not resolve its role in pathogenic adaptation and speciation of the M. haemolytica + M. glucosida subclade. Although genes and accessory elements gained from distantly related donors are responsible for many interesting adaptations of bacteria, other evolutionary processes are by no means inconsequential for adaptation to new ecological niches: (i) fixation of favorable mutations via selective sweeps or population bottlenecks; (ii) homologous replacement of alleles that have evolved in ecologically distant donors; or (iii) genome rearrangements (deletions, duplications, translocations, and inversions) via homologous recombination between direct and inverted repeats.
Previous works have revealed the relevance of homologous recombination between inter- and intragenera species in shaping the lkt operon (Davies et al. 2001, 2002). Our results are compatible with the predictions of Davies et al. (2001, 2002) on the mosaic origin of the lkt operon from M. haemolytica, M. glucosida biogroup 3B, and [P.] trehalosi, thus reaffirming the hypothesis that these gene transfer events could have important implications for pathogenic adaptation and speciation of the recipients. Indeed we have found a number of strains to be in incongruent positions on the rtxA tree (e.g., M. varigena strains 177T, V1835, and H39 group with M. glucosida strain P730 and M. granulomatis strain Ph13). Since these potential gene transfer events do not result in an incongruent position of the involved Mannheimia strains out from this genus, it seems reasonable to assume that they have occurred among strains belonging to genus Mannheimia via homologous replacement of vertically inherited +845/+1302 regions and not with other more distantly related taxa. However, our rtxA data set only included 458 bp after removal of ambiguous bases. In consequence, we need to address directly the question of the origin of each region by extending the data set of Davies et al. (2002) with genes from more distantly related taxa in order to explain the evolution of the entire lkt operon in light of homologous replacement of sequences outside the +845/+1322 region.
Conclusions
After the radiation from their common ancestor, the Mannheimia subclades have taken divergent paths toward their distinct lifestyles. However, we found no evidence for HGT of the lkt operon into any Mannheimia subclades, suggesting that alternative processes are involved in pathogenic adaptation and speciation. By contrast, the results presented here suggest that losses of the lkt operons in the radiating lineages of M. ruminalis have catalyzed their adaptation to a commensal environment and reproductive isolation (speciation).
References
Akaike H (1973) Information theory as an extension of the maximum likelihood principle. In: Petrov BN, Csaksi F (eds) 2nd International Symposium on Information Theory. Akademiai Kiado, Budapest, pp 267–281
Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Ambagala TC, Ambagala AP, Srikumaran S (1999) The leukotoxin of Pasteurella haemolytica binds to β2 integrins on bovine leukocytes. FEMS Microbiol Lett 179:161–167
Andersson JO, Andersson SG (2001) Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol Biol Evol 18:829–839
Angen Ø, Olsen JE, Bisgaard M (1997a) Further studies of the relationships among strains classified as taxon 15, taxon 18, taxon 20, [Pasteurella] granulomatis of the [Pasteurella] haemolytica-complex in ruminants using quantitative evaluation of phenotypic data. Zentralbl Bakteriol 286:317–332
Angen Ø, Aalbaek B, Falsen E, Olsen JE, Bisgaard M (1997b) Relationships among strains classified with the ruminant Pasteurella haemolytica-complex using quantitative evaluation of phenotypic data. Zentralbl Bakteriol 285:459–479
Angen Ø, Caugant DA, Olsen JE, Bisgaard M (1997c) Genotypic relationships among strains classified under the [Pasteurella] haemolytica-complex as indicated by ribotyping and multilocus enzyme electrophoresis. Zentralbl Bakteriol 286:333–354
Angen Ø, Mutters R, Caugant DA, Olsen JE, Bisgaard M (1999) Taxonomic relationships of the [Pasteurella] haemolytica complex as evaluated by DNA-DNA hybridizations and 16S rRNA sequencing with proposal of Mannheimia haemolytica gen. nov., comb. nov., Mannheimia granulomatis comb. nov., Mannheimia glucosida sp. nov., Mannheimia ruminalis sp. nov. and Mannheimia varigena sp. nov. Int J Syst Bacteriol 49:67–86
Blackall P, Angen Ø, Fegan N, Blackall L, Mutters R, Bisgaard M (2001) Characterisation of a novel Mannheimia sp from Australian feedlot cattle. Aust Vet J 79:634–639
Boucher Y, Douady CJ, Papke RT, Walsh DA, Boudreau MER, Nesbø CL, Case RJ, Doolittle WF (2003) Lateral gene transfer and the origins of prokaryotic groups. Annu Rev Genet 37:283–328
Burnham KP, Anderson DR (2002) Model selection and multi-model inference: a practical information-theoretic approach. Springer–Verlag, New York
Burrows LL, Olah-Winfield E, Lo RY (1993) Molecular analysis of the leukotoxin determinants from Pasteurella haemolytica serotypes 1 to 16. Infect Immun 61:5001–5007
Campbell A, Mrazek J, Karlin S (1999) Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA 96:9184–9189
Chang YF, Renshaw HW, Martens RJ, Livingston CW Jr (1986) Pasteurella haemolytica leukotoxin: chemiluminescent responses of peripheral blood leukocytes from several different mammalian species to leukotoxin- and opsonin-treated living and killed Pasteurella haemolytica and Staphylococcus aureus. Am J Vet Res 47:67–74
Christensen H, Kuhnert P, Olsen JE, Bisgaard M (2004) Comparative phylogenies of the housekeeping genes atpD, infB and rpoB and the 16S rRNA gene within the Pasteurellaceae. Int J Syst Evol Microbiol 54:1601–1609
Cooper VS, Lenski RE (2000) The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407:736–739
Davies RL, Whittam TS, Selander RK (2001) Sequence diversity and molecular evolution of the leukotoxin (lktA) gene in bovine and ovine strains of Mannheimia (Pasteurella) haemolytica. J Bacteriol 183:1394–1404
Davies RL, Campbell S, Whittam TS (2002) Mosaic structure and molecular evolution of the leukotoxin operon (lktCABD) in Mannheimia (Pasteurella) haemolytica, Mannheimia glucosida, and Pasteurella trehalosi. J Bacteriol 184: 266–277
Deshpande MS, Ambagala TC, Ambagala AP, Kehrli ME Jr, Srikumaran S (2002) Bovine CD18 is necessary and sufficient to mediate Mannheimia (Pasteurella) haemolytica leukotoxin-induced cytolysis. Infect Immun 70:5058–5064
Felsenstein J (2003) Inferring phylogenies. Sinauer Associates, Sunderland, MA
Highlander SK, Chidambaram M, Engler MJ, Weinstock GM (1989) DNA sequence of the Pasteurella haemolytica leukotoxin gene cluster. DNA 8:15–28
Highlander SK, Engler MJ, Weinstock GM (1990) Secretion and expression of the Pasteurella haemolytica leukotoxin. J Bacteriol 172:2343–2350
Highlander SK, Fedorova ND, Dusek DM, Panciera R, Alvarez LE, Rinehart C (2000) Inactivation of Pasteurella (Mannheimia) haemolytica leukotoxin causes partial attenuation of virulence in a calf challenge model. Infect Immun 68:3916–3922
Hooper SD, Berg OG (2002) Detection of genes with atypical nucleotide sequence in microbial genomes. J Mol Evol 54:365–375
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jackman TR, Larson A, de Queiroz K, Losos JB (1999) Phylogenetic relationships and tempo of early diversification in Anolis lizards. Syst Biol 48:254–285
Jeyaseelan S, Hsuan SL, Kannan MS, Walcheck B, Wang JF, Kehrli ME, Lally ET, Sieck GC, Maheswaran SK (2000) Lymphocyte function-associated antigen 1 is a receptor for Pasteurella haemolytica leukotoxin in bovine leukocytes. Infect Immun 68:72–79
Jeyaseelan S, Kannan MS, Hsuan SL, Singh AK, Walseth TF, Maheswaran SK (2001) Pasteurella (Mannheimia) haemolytica leukotoxin-induced cytolysis of bovine leukocytes: role of arachidonic acid and its regulation. Microb Pathog 30:59–69
Kaehler KL, Markham RJ, Muscoplat CC, Johnson DW (1980) Evidence of species specificity in the cytocidal effects of Pasteurella haemolytica. Infect Immun 30:615–616
Karlin S, Mrazek J, Campbell AM (1997) Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol 179:3899–3913
Koonin EV, Makarova KS, Aravind L (2001) Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol 55:709–742
Lally ET, Kieba IR, Sato A, Green CL, Rosenbloom J, Korostoff J, Wang JF, Shenker BJ, Ortlepp S, Robinson MK, Billings PC (1997) RTX toxins recognize a β2 integrin on the surface of human target cells. J Biol Chem 272:30463–30469
Lawrence JG (1997) Selfish operons and speciation by gene transfer. Trends Microbiol 5:355–359
Lawrence JG (1999) Gene transfer, speciation, and the evolution of bacterial genomes. Curr Opin Microbiol 2:519–523
Lawrence JG (2002) Gene transfer in bacteria: speciation without species? Theor Popul Biol 61:449–460
Lawrence JG, Ochman H (1997) Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol 44:383–397
Lawrence JG, Roth JR (1996) Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics 143:1843–1860
Lawrence JG, Roth JR (1999) Genomic flux: genome evolution by gene loss and acquisition. In: Charlebois RL (ed) Organization of the prokaryotic genome. ASM Press, Washington, DC, pp 263–289
Leite F, Sylte MJ, O’Brien S, Schultz R, Peek S, van Reeth K, Czuprynski CJ (2002) Effect of experimental infection of cattle with bovine herpesvirus-1 (BHV-1) on the ex vivo interaction of bovine leukocytes with Mannheimia (Pasteurella) haemolytica leukotoxin. Vet Immunol Immunopathol 84:97–110
Li J, Clinkenbeard KD, Ritchey JW (1999) Bovine CD18 identified as a species specific receptor for Pasteurella haemolytica leukotoxin. Vet Microbiol 67:91–97
Lo RY, Strathdee CA, Shewen PE (1987) Nucleotide sequence of the leukotoxin genes of Pasteurella haemolytica A1. Infect Immun 55:1987–1996
Mira A, Ochman H, Moran NA (2001) Deletional bias and the evolution of bacterial genomes. Trends Genet 17:589–596
Moran NA, Mira A (2001) The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol 2:research0054
Morgenstern B (1999) DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15:211–218
Murphy GL, Whitworth LC, Clinkenbeard KD, Clinkenbeard PA (1995) Hemolytic activity of the Pasteurella haemolytica leukotoxin. Infect Immun 63:3209–3212
Nilsson AI, Koskiniemi S, Eriksson S, Kugelberg E, Hinton JC, Andersson DI (2005) Bacterial genome size reduction by experimental evolution. Proc Natl Acad Sci USA 102:12112–12116
Nylander JA (2004) MrModeltest, version 2.2. Uppsala University, Uppsala
Petras SF, Chidambaram M, Illyes EF, Froshauer S, Weinstock GM, Reese CP (1995) Antigenic and virulence properties of Pasteurella haemolytica leukotoxin mutants. Infect Immun 63:1033–1039
Posada D, Buckley TR (2004) Model selection and model averaging in phylogenetics: advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808
Rambaut A, Drummond AJ (2004) Tracer, version 1.3. University of Oxford, Oxford
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schluter D (2000) The ecology of adaptive evolution. Oxford University Press, Oxford
Shewen PE, Wilkie BN (1982) Cytotoxin of Pasteurella haemolytica acting on bovine leukocytes. Infect Immun 35:91–94
Silva FJ, Latorre A, Moya A (2001) Genome size reduction through multiple events of gene disintegration in Buchnera APS. Trends Genet 17:615–618
Slowinski JB (2001) Molecular polytomies. Mol Phylogenet Evol 19:114–120
Strathdee CA, Lo RY (1989) Regulation of expression of the Pasteurella haemolytica leukotoxin determinant. J Bacteriol 171:5955–5962
Tatum FM, Briggs RE, Sreevatsan SS, Zehr ES, Hsuan SL, Whiteley LO, Ames TR, Maheswaran SK (1998) Construction of an isogenic leukotoxin deletion mutant of Pasteurella haemolytica serotype 1: characterization and virulence. Microb Pathog 24:37–46
Vulic M, Dionisio F, Taddei F, Radman M (1997) Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria. Proc Natl Acad Sci USA 94:9763–9767
Vulic M, Lenski RE, Radman M (1999) Mutation, recombination, and incipient speciation of bacteria in the laboratory. Proc Natl Acad Sci USA 96:7348–7351
Welch RA (2001) RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr Top Microbiol Immunol 257:85–111
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z, Roberts D (1995) On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol Biol Evol 12:451–458
Zhang J (1999) Performance of likelihood ratio tests of evolutionary hypotheses under inadequate substitution models. Mol Biol Evol 16:868–875
Acknowledgments
We thank Christel G. Buerholt and Tony Bønnelycke for technical assistance and Sean D. Hooper for enlightening discussions. This work was supported by Grant 9702797 from the Danish Agricultural and Veterinary Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. David Guttman]
Jesper Larsen and Anders G. Pedersen contributed equally to this work.
Rights and permissions
About this article
Cite this article
Larsen, J., Pedersen, A.G., Christensen, H. et al. Evidence for Vertical Inheritance and Loss of the Leukotoxin Operon in Genus Mannheimia . J Mol Evol 64, 423–437 (2007). https://doi.org/10.1007/s00239-006-0065-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0065-3