
Abstract
FimH, the mannose-specific, type 1 fimbrial adhesin of Escherichia coli, acquires amino acid replacements adaptive in extraintestinal niches (the genitourinary tract) but detrimental in the main habitat (the large intestine). This microevolutionary dynamics is reminiscent of an ecological “source-sink” model of continuous species spread from a stable primary habitat (source) into transient secondary niches (sink), with eventual extinction of the sink-evolved populations. Here, we have adapted two ecological analytical tools—diversity indexes D S and α—to compare size and frequency distributions of fimH haplotypes between evolutionarily conserved FimH variants (“source” haplotypes) and FimH variants with adaptive mutations (putative “sink” haplotypes). Both indexes show two- to threefold increased diversity of the sink fimH haplotypes relative to the source haplotypes, a pattern that ran opposite to those seen with nonstructural fimbrial genes (fimC and fimI) and housekeeping loci (adk and fumC) but similar to that seen with another fimbrial adhesin of E. coli, papG-II, also implicated in extraintestinal infections. The increased diversity of the sink pool of adhesin genes is due to the increased richness of the haplotypes (the number of unique haplotypes), rather than their evenness (the extent of similarity in relative abundances). Taken together, this pattern supports a continuous emergence and extinction of the gene alleles adaptive to virulence sink habitats of E. coli, rather than a one-time change in the habitat conditions. Thus, ecological methods of species diversity analysis can be successfully adapted to characterize the emergence of microbial virulence in bacterial pathogens subject to source-sink dynamics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the course of spread of a microbial population from one habitat to another, it undergoes niche differentiation that may involve adaptive functional changes of individual genes (Orr and Smith 1998). However, due to limited numbers of such adaptive mutations and their variable nature, it can be difficult to recognize genes targeted by adaptive mutations. This is especially difficult during the early stages of niche differentiation, when adaptive mutations in a gene under selection occur multiple times in different strains in diverse haplotype backgrounds. Independent mutational changes in diverse haplotypes may create a pattern of variation that is not substantially different from one created by accumulation of selectively neutral mutations. Thus, adaptive and neutral variability may not be distinguished by conservative methods of molecular evolutionary analysis, such as the ratio of nonsynonymous substitution rate to synonymous substitution rate (d N /d S ) (Nei and Gojobori 1986), Tajima’s (1989) D, and or Fu and Li’s (1993) D* statistics (Sokurenko et al. 2004). We analyze here whether ecological diversity indexes can be used to compare the allelic distributions in haplotype sets (from the same bacterial strains) of genes believed to be under either long-term neutral or, alternatively, short-term positive selection, with an aim to identify candidate genes accumulating adaptive mutations in the course of niche differentiation.
We have previously described a method, termed zonal phylogeny (ZP) analysis, where an unrooted protein phylogram is built from the corresponding DNA phylogram, distinguishing two categories of protein variants—those encoded by multiple unique haplotypes (i.e., alleles differing only by synonymous changes) and those encoded by a single haplotype (Sokurenko et al. 2004). All nodes composed of multihaplotype variants are combined into a Primary zone of the tree, while nodes composed of monohaplotype variants are combined into an External zone. The Primary zone proteins thus represent evolutionarily stable structural variants that have circulated over long evolutionary time, with the coding alleles gradually accumulating silent changes. In contrast, the External zone proteins represent variants that have evolved relatively recently, without silent site variation having yet accumulated.
In a previous study (Sokurenko et al. 2004), we used ZP to detect selection footprints in the Escherichia coli gene encoding mannose-specific type 1 fimbrial adhesin, FimH. FimH is a lectin-like protein (30 kDa) located on the fimbrial tip (Klemm and Christiansen 1987). Naturally occurring point replacements in various locations throughout the FimH protein increase its ability to bind monomannose (1M) receptors on uroepithelial cells, and such mutant alleles are common among uropathogenic, but not fecal, isolates (Sokurenko et al. 1995, 1997). It was shown that alleles from the External zones of the FimH tree have derived recently from Primary zone alleles, which we hypothesize could be explained by source-sink evolutionary dynamics in fimH. Under the source-sink model, the genetic lineages forming the Primary zone nodes circulate in an evolutionarily stable “source” niche for E. coli—the large intestine of healthy mammals. These bacteria, however, are continuously spreading into extraintestinal compartments (such as the urinary tract) where selection results in the adaptive mutation of source fimH to increase bacterial tropism to uroepithelium (Sokurenko et al. 1998; Hommais et al. 2003). These mutations, however, appear to be deleterious in the original intestinal niche, due to a functional trade-off: 1M-enhancing mutations also produce heightened sensitivity to inhibition of binding by mannosylated glycoproteins present in soluble form in saliva and intestinal mucus. As urinary tract infections are acute and self-resolving in nature, urinary tract habitats are transient for E. coli (i.e., they represent “sink” niches). When bacteria with the uro-adapted FimH return from the alternative niche into fecal-oral circulation in the primary intestinal habitat, they are outcompeted by bacteria expressing the conserved low 1M-binding, but less inhibitable, FimH variants. Thus, mutant fimH alleles cannot sustain the uro-adapted clones of E. coli in the long term. However, invasion of new strains from the reservoir into the urinary tract constantly selects for new mutant strains and, thus, constitutes a continuous pool of recently-evolved “sink” FimH variants.
The source-sink scenario is a novel model of bacterial gene dynamics and could represent a major mode of virulence evolution (Sokurenko et al. 2006). Source-sink was originally developed as an ecological model (Pulliam 1988) and has not yet been described from the molecular evolutionary perspective. Here, to gain insight into the source-sink dynamics of E. coli microevolution, we used ecological diversity indexes to compare the haplotype size (i.e., the number of sampled organisms carrying a particular haplotype) and the distribution of frequencies of haplotypes of a particular size of five genetic loci from intestinal and uropathogenic strains of E. coli—fimH, fimC (encoding the molecular chaperone of type 1 fimbriae), fimI (encoding a putative regulator of type 1 fimbrial biogenesis), papG-II (encoding the fimbrial adhesin of di-galactose-specific P fimbriae subclass II), and two housekeeping genes, adk and fumC (encoding adenine kinase and fumarase C, respectively).
Methods
Strains Analyzed
The datasets of adk, fumC, fimC, and fimI genes included the same 75 strains, of which there were 25 fecal, 30 urinary (10 cystitis, 10 pyelonephritis, 10 urosepsis), and 20 non-urinary tract infection (UTI)-associated extraintestinal isolates (5 from sputum, 4 of wound origin, and 11 bacteremia strains). The papG-II dataset had 68 sequences from 67 strains (CFT073 having two papG-II genes), carrying 6 fecal, 34 urinary (5 cystitis, 10 pyelonephritis, 19 urosepsis), and 27 non-UTI extraintestinal (3 bacteremia, 23 vaginal) isolates. The strains were collected from different parts of the United States, the Netherlands, Benin, Denmark, Kenya, and Sweden, and encompassed a wide range of serotypes, including representatives of canonical extraintestinal pathogenicity-associated antigens O1, O2, O4, O6, O7, and O18. Eighteen strains were common to both datasets.
Zonal Phylogeny (ZP) Analysis
For each gene, an unrooted ML DNA tree was constructed, based on a general, time-reversible model (PAUP* 4.0b). Rates of substitution were estimated from the sequence data. To increase computing time efficiency, the input sample set included only the unique haplotypes (alleles).
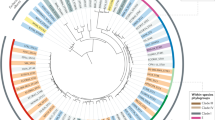
In Fig. 1, the tree nodes correspond to unique haplotypes in the sample set, with node size representing the frequency of each haplotype. The tree nodes were divided into two zones—the internal Primary zone and the External zone—based on whether or not (respectively) a given haplotype differed from any other haplotype in the sample set only by synonymous changes. Therefore, all sequences encoding multihaplotype protein variants (i.e., those encoded by more than one allele differed by silent variability only) are assigned to the Primary zone. Any sequence positioned between two Primary zone haplotypes (regardless of the nature of its variation from either Primary node) is also assimilated into the Primary zone. The External zone encompasses the remaining haplotypes, each differing from all other haplotypes in the dataset by at least one nonsynonymous mutation. Thus, the External zone is composed of haplotypes encoding monohaplotype protein variants.

Unrooted maximum-likelihood DNA trees for adk, fumC, fimC, fimI, fimH, and papG-II genes from E. coli strains. Each node represents a unique allele (haplotype), with node size corresponding to number of strains (haplotype size). Branch lengths do not reflect the number of corresponding nucleotide changes. The internal Primary zone is separated from the External zone by the border of thick dashed line. Each dotted branch represents a synonymous substitution(s), while each solid branch includes at least one nonsynonymous substitution.
Calculation of D S
Simpson’s (1949) index measures the probability that two individuals chosen independently and at random from a large heterogeneous species population will be found to belong to the same species, and is given by
where S is the total number of species, π i is the relative frequency of the ith species, and i goes from 1 to S. This formula suggests that the greater the value of λ, the greater the probability that any two individuals belong to the same species, the more uneven the species-abundance distribution, and, in effect, the less diverse the population. The inverse of Simpson’s index (Hill 1973) is taken to form the diversity index, D S :
The diversity measure D S increases with both the evenness of relative abundances of different species and the richness of species in the ecological zone under study.
For very large N, the total number of individuals in the sample, the variance of λ was approximated by Simpson (1949) as
In our case, species are replaced by haplotypes; therefore, S becomes the total number of haplotypes, π i is the relative frequency of the ith haplotype and N represents the total number of strains in a particular zone. For each gene, we measured D S separately for the two zones, Primary and External, and then used z-statistics to calculate the level of significance for differences between the zonewise values.
Calculation of α
The distribution of the number of individuals sampled independently from homogeneous material (e.g., consisting of numerous individuals of a single species) follows a Poisson series, given by the formula
where n is the number of observed individuals in any sample and m represents the expected number, the average value of n. However, when the sampled material is heterogeneous (e.g., a collection of different species, each represented by a number of individuals), there would be a mixture of distributions for different values of m. From such a heterogeneous population, the probability of observing the number n in the samples is given by
which is related to the negative binomial expansion, as given by
and therefore is called a negative binomial distribution (Fisher 1943; White and Bennetts 1996) and is a natural extension of (4). The parameter p is proportional to the sample size, and the expectation or the average value of n is pk. k is an inverse measure of variability of the different expectations of the component Poisson series. For very large values of k, expectations are nearly equal and the distribution tends toward Poisson form, suggesting homogeneity in the samples. On the contrary, as k approaches its limiting value 0, the samples represent greater heterogeneity.
Now as we modify (5), setting k = 0, replacing p/(1 + p) by x (so that 0 < x < 1, varying with the sample size), and replacing the constant factor (k – 1)! in the denominator by a new constant factor α in the numerator, the simplified expression for the expected number of species with n individuals becomes
where the index α represents a direct measure of the extent of variability in the sample.
Consequently, the total number of species is given by
and similarly, the total number of individuals is given by
The two relationships in (8) and (9) allow us to determine the value of α, given values of S and N. To compare the different estimates of α, we need the standard error of the α values; the variance of α is shown (Fisher 1943) to be approximately
We used this ecological index of variability (Pielou 1969) to compare the haplotype size frequencies in the Primary and External zones of the DNA trees for each of six genes of interest. However, in our case, the number of different species (S) is synonymous with the number of different haplotypes, while the total number of individuals in the sample population (N) means here the total number of bacterial strains. For each gene in this study, we calculated two α values separately for the two zones, based on the corresponding values of S and N; the significance level of their difference was calculated using z-statistic.
Intergene Comparison of Differential Zonal Diversity
For each gene, we computed the difference in the level of haplotype diversity between the Primary (P) and the External (E) zones in terms of the difference in λ values (Eq. 1) and α values (solved from Eqs. 8 and 9) as (λ E –λ P ) and (α E –α P ), respectively. The z-statistic was applied to identify significant comparisons of differential zonal haplotype diversity between gene pairs.
The Evenness Factor
D S , the inverse of Simpson’s index λ, is considered as the effective number of species in the sample population of an ecological zone, and is a direct measure of diversity, incorporating the factors of species richness (S, the observed number of species) and evenness. Though we did not explicitly know how the species richness factor and the evenness factor were intermingled in the overall diversity measure, we tried here to minimize the species (or, in our case, haplotype) richness factor by using the ratio of the effective number to the observed number of species (haplotypes) as a crude measure of the evenness factor, given by
where the value ranges from 0 to 1. In the present case, the higher this ratio in a haplotype zone, the more even (and more diverse) the haplotype population.
Ewens-Watterson Homozygosity Test
We used PyPop 0.6.0 (Lancaster et al. 2003) to perform the Ewens-Watterson homozygosity test of neutrality (Ewens 1972; Watterson 1978; Slatkin 1994, 1996). It computes the observed homozygosity (F obs ) as the sum of the squares of haplotype frequencies. The expected homozygosity under neutrality (F exp ), for the same sample size and number of unique haplotypes, is obtained by simulation. The normalized deviate of the homozygosity (F nd ) is calculated as the difference between the F obs and F exp , divided by the square root of the variance of F exp (Salamon et al. 1999). Negative F nd implies an observed homozygosity level lower than expected homozygosity, in the direction of balancing selection when there is significant deviation from neutrality expectation. A significant positive value is indicative of directional selection. The p-value of F is the probability that the observed homozygosity would be obtained from a neutral sample (of identical sample size and identical number of distinct haplotypes). To make this a two-tailed test of the null hypothesis of neutrality, the p-values at either extreme of the distribution are considered significant: p F < 0.025 or p F > 0.975 significant at the 0.05 level, and p F < 0.05 or p F > 0.95 significant at the 0.10 level.
Results
Haplotype Distribution in ZP Tree Zones
Each gene tree is presented as a diagrammatic, unrooted maximum likelihood (ML) DNA cladogram (Fig. 1) with Primary and External zones that combine multi- and monohaplotype nodes, respectively (as described under Methods). In the fimC, fimI, fimH, and papG-II trees, the Primary zone had its position in the center of the cladogram, consistent with Primary haplotypes being evolutionarily original to haplotypes in the External zone (Fig. 1). In the housekeeping gene trees (adk and fumC), however, such relationships were difficult to define due to comparatively poor development of the External zones. The distribution of the numbers of haplotypes of particular size in the Primary and External zones is presented for each gene in Table 1. By comparing the frequency of fecal strains, urinary tract infection (UTI)-causing strains (combining strains of cystitis, pyelonephritis, and urosepsis origin) and non-UTI extraintestinal pathogenic strains in the Primary and External zones of each gene (Table 2), we found significant predominance of UTI strains compared to fecal strains in the External zone for fimH (p = 0.03), unlike for the housekeeping genes, fimI and fimC. In contrast, P fimbriae are not ubiquitous like type 1 fimbriae but, rather, have entered into select E. coli lineages, as part of “pathogenicity-associated islands” that promote extraintestinal virulence (Welch et al. 2002). Table 2 shows a significant predominance of UTI strains compared to non-UTI extraintestinal ones in the papG-II External zone (p = 0.04), supporting the association between the carriage of P fimbriae and adaptive urovirulence (Roberts et al. 1994). However, the ratio of Primary zone-to-External zone haplotypes of papG-II was similar for fecal strains and UTI strains, with the External zone quite well developed for both, though the overall fecal sample size for papG-II was considerably low. The similarity in ratios may indicate that the intestinal niche serves as a habitat for P-fimbriated bacteria where the papG-II diversification is selected; thus the intestinal niche can be a sink habitat similar to the urinary tract, while a non-urinary tract niche (for instance, the vaginal introitus) serves as the source habitat. Alternately, the fecal strain External zone may reflect oversampling of urinary tract-adapted strains circulating back into the intestinal niche.
To determine whether the differential distribution of fecal and UTI isolates in the tree is a result of positive selection on the latter, we calculated the d N /d S value for each set, based on a null model of nearly neutral selection and a model of positive selection, using PAML v3.15 (Yang 1997). The likelihood ratio test did not show any significant difference between the two models for any of the datasets. The average d N /d S value for the entire fimH dataset, calculated, was well below 1 (0.18), with the UTI-only dataset giving a slightly higher d N /d S value (0.16) than the fecal-only dataset (0.09). Though this trend is consistent with the observed predominance of UTI strains in the External zone of the fimH zonal phylogeny (Table 2), the overall low level of the values provides no support for the presence of selection. The lack of support, however, could be due to the low sensitivity of the PAML analysis when applied to this type of dataset. Therefore, we decided to try another approach for the analysis.
We hypothesized that if haplotypes in the Primary and External zones were formed under neutral and positive selection, respectively, haplotype diversity in the External zone would be different from the Primary zone, and the nature of this difference would depend on the type of selection in the External zone.
Haplotype Diversity Based on the DS Index
To determine whether there were differences between the Primary and the External zones in both size and frequency of haplotypes, we opted for the diversity measure D S . As a reciprocal of Simpson’s index λ, D S is widely used to measure species diversity in an ecological habitat based on species richness (total number of species) and evenness (extent of similarity in relative abundance of different species). For this analysis, we treated (i) the Primary and the External zones for each gene as two separate and independent ecological habitats (a reasonable assumption inasmuch as the functional mutations originate independently from each other and independent of allelic background); (ii) each haplotype as a particular species; (iii) specific haplotype size as the frequency of individuals representing a particular species; and (iv) the frequency of haplotypes of a particular size as equivalent to the frequency of species represented by a particular number of individuals. D S , as the inverse of λ, increases with increasing diversity, which, in turn, directly correlates with the species (haplotype) richness and evenness.
For adk, fumC, fimC and fimI, the Primary zone haplotype diversity values well exceeded the corresponding values of the External zone haplotypes (Fig. 2). For fimH and papG-II, the picture was reversed: the fimH External zone value was not only larger than the Primary zone value, but larger than all zone values for the other four genes, while its Primary zone diversity value remained in the range of Primary zone values for the other genes. However, if we ignore the Primary-External diversity comparisons for the two housekeeping genes (which might not be reliable due to small sample sizes in the External zones of adk and fumC), the difference between zones for a given gene was statistically significant only for papG-II (p < 0.05).

The D S values for the Primary (P) and External (E) zones of six genes. Vertical bars denote standard errors.
In contrast to papG-II, the haplotype diversity of the fimH External zone was not statistically different from that of the fimH Primary zone, according to the D S index. However, we hypothesized that, similar to papG-II, the upward shift in the D S value for fimH External zone haplotypes could be significant compared to the zonal differences in other genes. Therefore, we computed the differences of λ values of the External and Primary zones as (λ E – λ P ) for all genes. (λ is used here in place of D S for ease of calculation, and it should be kept in mind that λ is inversely proportional to the increase in diversity measured by D S .) As expected, the genes with negative values for (λ E – λ P ) are fimH and papG-II (Fig. 3). The fimH (λ E – λ P ) value was significantly different from those of adk (p = 0.011), fumC (p = 0.002), and fimC (p = 0.03); however, fimH (λ E – λ P ) did not differ significantly from the fimI value (p = 0.105) or from 0 (p = 0.111). In addition to fimH, fimI (λ E – λ P ) also deviated significantly from the adk (p = 0.035) and fumC (p = 0.018) values. The difference of fimC (λ E – λ P ) from the adk and fumC values approached the significance threshold (p = 0.067 and p = 0.065, respectively). On the other hand, papG-II (λ E – λ P ) deviated significantly from all the rest: adk (p < 0.001), fumC (p < 0.001), fimC (p < 0.001), fimI (p = 0.002), fimH (p = 0.026), and 0 (p = 0.002).

The differences in Simpson index (λ) values for the External and Primary zones, given as (λ E – λ P ). Vertical bars denote standard errors.
Thus, the D S index indicated that the diversity of fimH and papG-II External zone haplotypes might be greater than that of corresponding Primary zone haplotypes. Moreover, the External zone haplotype diversity in fimH exceeded the diversity of haplotypes in any other zone for the rest of the genes analyzed, though the differences were not statistically significant. However, the (λ E –λ P ) calculation, which measures the zonal diversity differential for each gene, demonstrated a significant increase in the diversity of fimH External zone haplotypes. At the same time, papG-II (λ E –λ P ) showed the highest relative increase in External zone haplotype diversity, significantly different from the other five genes.
Haplotype Diversity Based on the α Index
In order to increase statistical power for detecting haplotype diversity trends, we moved to a model-based (but analytically more complex) diversity index, α. Rather than incorporating frequency values for each haplotype, the α model assumes that the abundance for each haplotype follows a Poisson distribution, which we believe is a reasonable assumption in our case (see Discussion). Thus, we used the values of S (number of unique haplotypes) and N (number of strains) for each zone in all six genes to determine the values of α (Fig. 4). The trend in diversity distribution with this approach was similar to what we found for D S : for all genes except fimH and papG-II, there was higher variability in the Primary zone than in the External zone. The fimH Primary zone α value was in the range of Primary zone values for fimI and fimC, while the External zone α value was significantly higher. In contrast to D S , the statistical comparisons between α values reveal significant differences in haplotype frequency distribution variability for fimH External zone haplotypes compared to fimH Primary zone haplotypes (p = 0.03), as well as to the haplotypes from the adk Primary (p = 0.004), fumC Primary (p = 0.01), fimC Primary (p = 0.054) and External (p = 0.009), fimI Primary (p = 0.04) and External (p = 0.005), and papG-II Primary (p < 0.001) zones. In the case of papG-II, the Primary zone α value was the lowest of all α values determined here, but not statistically different from α values for any other gene, except for adk Primary zone (p = 0.02). (We did not include the External α values for adk and fumC in the statistical comparisons due to small sample size.) However, the External zone α value of papG-II was significantly higher than the corresponding Primary zone value (p < 0.001). The only other significant difference in zonal diversity was found between fimC Primary and adk Primary (p = 0.03).

The α values for the Primary (P) and External (E) zones of six genes. Vertical bars denote standard errors.
The D S and α indexes were clearly related (Fig. 5), and the linear regression analysis gave a good fit (R = 0.86). Curvilinear relationship gave even a slightly better fit (not shown), since the relationship was not strictly linear. At lower levels of diversity, D S and α tracked closely, but at higher levels of diversity, α values increased more rapidly than D S . In the case of the fimH External zone—the most diverse of the zones studied here—α became almost double D S . The reasons for the rapid increase in α relative to D S were difficult to ascertain, as we were not aware of an explicit functional relationship between α (as a parameter of the negative binomial distribution) and D S (or λ, from which D S is derived). At the same time, the more rapid increase in α at higher levels of diversity provided improved resolution of (α E – α P ) values (Fig. 6) relative to (λ E – λ P ) values as a measure of differential zonal haplotype diversity. Specifically, the (α E – α P ) values for fimH and papG-II represented the only two positive values among the six genes of interest, and were significantly different from 0 (p = 0.03 and p < 0.001, respectively) and from the (α E – α P ) of the remaining genes—adk (p = 0.014 and p < 0.001), fumC (p = 0.005 and p < 0.001), fimC (p = 0.011 and p < 0.001), and fimI (p = 0.012 and p < 0.001)—though not significantly different from each other (p = 0.67). At the same time, no pairwise (α E – α P ) comparisons between any non-fimH genes were significant.

Plot of D S vs. α for both the Primary and the External zones of six genes. The linear fit of the plot is shown (R = 0.86).

Differences in the α values for the External and Primary zones, given as (α E – α P ). Vertical bars denote standard errors.
To assess the possible role of recombination in creating excessive haplotype diversity in the fimH External zone, we removed an approximately 220-bp region within fimH that had shown a high level of recombination (unpublished observations). Analysis of the modified fimH haplotypes revealed a pattern of diversity similar to the pattern obtained with the full-size fimH (not shown), suggesting that intragenic recombination was not responsible for creating excess fimH external zone diversity.
Thus, use of the α diversity index demonstrated that fimH and papG-II haplotypes from the External zone had a significantly higher level of diversity than Primary fimH and papG-II haplotypes, respectively, while the diversity level of fimH External zone haplotypes was significantly higher than that of haplotypes from either zone for the other genes studied, except for the External zone haplotypes of papG-II . The α statistic captured diversity information similar to that captured by D S , but in a more statistically sound way.
Haplotype Richness and Evenness Factors
The increase in fimH and papG-II External zone diversity could be due to the increase in one or both major parameters of the diversity indexes: haplotype richness (the number of unique haplotypes) and haplotype evenness (similarity in the size of different haplotypes). Richness (S) is used directly in the calculations of both D S and α, while evenness directly affects only D S and is equivalent to the D S /S ratio. Table 1 shows that the haplotype richness, S, for the fimH External zone was the highest of all the zones and was twice as high as the richness of the fimH Primary haplotypes. The opposite was true in the other genes except papG-II, where the External zone S was the second highest of all the zones and was almost eight times the Primary zone S. In contrast, the evenness values of the External zone haplotypes (given by D S /S; Table 1) were lower than the evenness of Primary zone haplotypes for fimH and papG-II but were higher for the other four genes analyzed. (Again, however, the evenness values in the External zones of adk and fumC could not be estimated reliably due to small sample size, producing values very close to 1, the maximum level of evenness.) Therefore, the increased diversity of fimH and papG-II haplotypes in the corresponding External zones was due to increased richness, rather than evenness.
Ewens-Watterson Test
Observed homozygosity (F obs ) values of any Primary zone haplotypes were not significantly different from the expected homozygosity value (F exp ) obtained for the haplotype distribution based on neutral evolution, as we computed F nd , the normalized difference between the two (Table 1), thus showing no sign of directional selection for the Primary zone alleles of any gene tested. F obs values of haplotypes from the External zones of fimI and fimC genes were also not different from the corresponding F exp , while the housekeeping genes External zone haplotypes were not considered in the analysis due to the negligible sample size. In contrast, F obs values for the fimH and papG-II haplotypes from External zone were significantly higher than obtained for the corresponding F exp (though for fimH the two-tailed significance was at the borderline level of 0.05 < p < 0.10, with the p F of normalized deviate value, F nd , being 0.968; see Methods). This suggested the presence of directional selection through the significant differences in fitness between bacterial clones carrying various fimH and papG-II alleles in the External zone. This result could arise from a combination of pathoadaptive alleles and slightly detrimental alleles in the external zone, but it also could signify that some pathoadaptive alleles are more fit overall than others. This was in concordance with the notion above that the increased diversity of External fimH and papG-II haplotypes was due to richness, not evenness.
Discussion
We present here the analysis of E. coli genes by zonal phylogeny (ZP), which separates protein variants into two basic categories, or zones: those encoded by multiple haplotypes (Primary zone) and those encoded by a single haplotype (External zone). We hypothesize that the multihaplotype variants have circulated in E. coli over long periods of time and are relatively well adapted to functioning in an evolutionary stable niche (“source” habitat), where they are under purifying selection against structural changes.
In contrast, monohaplotype protein variants likely represent recently derived variants that have not yet accumulated any silent variability. Though some of these variants may include rare variants that are under purifying selection, most of the monohaplotype variants appear to represent proteins that are under both positive and purifying selection, depending on the habitat. Our previous studies have shown that the FimH adhesin of E. coli adapts under positive selection through acquisition of point mutations that increase its monomannose-binding capability; this property is adaptive in extraintestinal niches of E. coli, such as the monomannose-rich epithelium of the urinary tract, but detrimental in circulation in the gastrointestinal habitat (Sokurenko et al. 1995, 1998, 2004), though no direct experimental evidence for the colonization trade-off has been provided yet. Colonization of the urinary bladder and kidney by E. coli generally manifests as an acute infection that naturally self-resolves within 1–2 weeks and is not typically transmitted from person to person. Thus, the urinary tract is a short-term transient habitat, indicating that it could be an unstable “sink” habitat for E. coli.
The hypothesis of a recent origin for External zone FimH variants conforms well to a “source-sink” model of E. coli microevolution (Sokurenko et al. 2004, 2006), under which bacteria continuously spread from an evolutionarily stable niche (source) into an alternative and relatively unstable habitat (sink). Under this scenario, a FimH mutation resulting in increased monomannose binding to surface-bound receptors is adaptive in the sink environment and may lead to the clonal expansion of E. coli there; at the same time, the mutation is detrimental in the original source habitat, due to a functional trade-off in which the mutated FimH is more sensitive to inhibition by soluble mannosylated glycoproteins abundant in gastrointestinal mucosa. Thus, the mutations that enhance monomannose binding by FimH will be eventually removed by selection over the long term. It is possible that upon return to the intestinal habitat, reversion mutations could occur in the urinary tract-adapted FimH variants, thus re-adapting the adhesin function to the source habitat. However, fimH mutations are unlikely to be the only sink-adaptive/source-detrimental changes accumulating in the course of alternative niche colonization. Assuming a high level of competition within the source habitat, we believe that the survival of sink-adapted clones via reversion of multiple mutations seems rather unlikely. However, to date, there are no published data to address this matter.
We show here that putative sink fimH haplotypes (comprising the External zone) have significantly higher diversity than source fimH haplotypes from the Primary zone. As the External zone haplotypes represent the monohaplotype FimH variants (most of which carry mutations adaptive for extraintestinal E. coli), the increased haplotype diversity could be a specific characteristic of genes involved in the adaptation to sink environments, under the source-sink evolutionary model. Indeed, haplotypes of fimC and fimI form External zones with much lower diversity than the corresponding Primary zone haplotypes. These two genes encode proteins involved in fimbrial biogenesis and their function does not have direct effects on the fimbrial receptor specificity, which is almost certainly the trait under selection in the course of niche adaptation. Not surprisingly, the diversity patterns of fimC and fimI are more similar to the patterns of the housekeeping genes adk and fumC, known to be under strong purifying selection against structural variation due to their importance in maintaining basic physiologic processes in the bacterial cell (Feil and Spratt 2001).
Interestingly, the diversity of Primary fimH haplotypes is very similar to the diversity of Primary haplotypes of fimC, fimI and housekeeping genes, indicating that they are subject to similar populational dynamics. However, fimH diversity exceeds that of the other genes in the corresponding External zones. We believe that this increase in the diversity of External zone fimH haplotypes is due to strong selective pressure for monomannose-enhancing FimH mutations in the sink habitat, and to the diverse nature and location (Weissman et al. 2006) of these mutations. This situation leads to the emergence of multiple adaptive variants from the same source haplotypes of the Primary zone, thus increasing the relative richness of the haplotypes in the External sink zone. Indeed, it is the increase in haplotype richness that is responsible for the increase in diversity of the External haplotypes, as the haplotype evenness has actually been reduced.
The statistical and biological significance of the decrease in evenness of the External zone fimH haplotypes is also supported by the Ewens-Watterson test, which shows that the adhesin genes exhibit more homogeneity (i.e., unevenness) in the External zone haplotypes than expected, based on the assumption of neutral evolution. The selection-driven, uneven distribution of fimH and papG-II alleles from the External zone could reflect their differential adaptive value. Indeed, structural mutations in FimH adhesin were shown to produce various degrees of the presumed uroadaptive functional change—the monomannose-binding capability that is directly correlated with the bacterial urotropism (Sokurenko et al. 1995). However, fimH haplotypes that form both the largest and the smallest (singleton) nodes in the External zone are commonly translated into structurally identical FimH variants (i.e., adaptively equivalent). This suggests that, despite the adaptive equivalency, these alleles might be carried by bacterial clones that are expanding and shrinking, respectively, in the population of bacterial pathogens, consistent with continuous emergence and extinction of bacterial clones under the source-sink model of pathogen evolution. The extinction of bacterial clones is expected to be caused by accumulation over time of multiple sink-adaptive mutations (throughout the genome) that impose increasing levels of functional trade-off back in the original source habitats. Thus, the increased unevenness of the adaptive alleles of FimH and PapG-II adhesins argues for ongoing source-sink dynamics of these bacterial pathogens, rather than for their emergence due to a recent, one-time change in the habitat.
On the other hand, the decrease in diversity of the External zone fimC, fimI and housekeeping genes haplotypes is due to a significant decrease in richness, compared to the Primary zone haplotypes, as the haplotype evenness of the former has in fact increased. The relative decrease in richness might reflect the fact that External zone haplotypes of the nonadhesin genes are not participating in the adaptive source-sink dynamics, but are the result of random genetic drift and might be (mildly) functionally deleterious compared to the Primary zone haplotypes. Thus, these External zone haplotypes could be subject only to purifying selection and, hence, be less diverse in haplotype richness—but more even in haplotype frequency distribution—than the well-adapted, stable haplotypes from the Primary zone. This situation may occur because selection is more efficient compared to drift as the frequency of mildly detrimental alleles increases.
As mentioned above, the source-sink model of fimH microevolution has been developed from ecological models of the same name (Pulliam 1988), which describe species spread from original to alternative environments. To characterize haplotype size and frequency distributions, we have successfully employed two diversity indexes (D S and α) that also originated from ecological studies; these studies evaluated the diversity of species populations, prey-predator relationships, and host-parasite relationships through randomly collected samples (Pielou 1969; Hill 1973). Thus, it appears that ecological models and statistics can be productively applied to the analysis of adaptive microevolutionary events on the level of single species.
The two indexes show identical trends in the levels of diversity for the Primary and External zones of six genes studied; however, the α index provided better statistical resolution. The D S is a straightforward, nonparametric, ad hoc diversity measure based on the calculation of relative abundances of all species in a sample (Hill 1973). In contrast, α is analytically complex and does not yield such an intuitive ecological meaning. Also, the α diversity calculation is based on a Poisson distribution fitted to observed species-abundance data, given the total number of individuals (strains) and number of different species (unique haplotypes) in the sample (Pielou 1969). Because the E. coli species exists as a diverse set of ecotypes, it is possible that random sampling of E. coli strains from specific habitats may be biased against some ecotypes and thus would not provide a Poisson distribution of haplotypes. However, one could argue that assumption of a Poisson distribution of haplotypes should not significantly disturb the α analysis, given that (i) E. coli ecotypes may not be entirely distinct from one another, either phylogenetically or environmentally; (ii) no specific lineages are associated specifically with urinary tract isolates; (iii) type 1 fimbrial clusters are subject to frequent horizontal transfer between different E. coli lineages (Weissman et al. 2006); and (iv) all genes compared in this study have derived from the same set of strains. Indeed, D S and α produce similar results and are not affected by sample size, although the latter index tends to provide better resolution. We also expect these indexes to demonstrate similar trends for the E. coli datasets of other ecotypes, including strains of nonhuman/nonanimal origin (e.g., found in environmental sites or agricultural products), depending on the differential stability of the alternative habitats.
Thus, we believe that both these indexes, as well as the (λ E – λ P ) and (α E – α P ) measures introduced here, could be used to track quantitatively the overall haplotype distribution diversity based on the relative abundances of unique haplotypes and the total number of unique haplotypes. In conjunction with the ZP analysis of DNA trees, these diversity indexes can be used to understand the microevolutionary dynamics of source-sink evolution of bacteria and, thus, the emergence of microbial virulence. This approach may have broader application. When enough multiple genomes of the same species accumulate, it could be used to identify candidate genes that are under short-term or source-sink selection.
References
Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Pop Biol 3:87–112
Feil EJ, Spratt BG (2001) Recombination and the population structures of bacterial pathogens. Annu Rev Microbiol 55:561–590
Fisher RA (1943) The relation between the number of species and the number of individuals in a random sample of an animal population. Part 3. A theoretical distribution for the apparent abundance of different species. J Anim Ecol 12:54–58
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Hill MO (1973) Diversity and evenness: a unifying notation and its consequences. Ecology 54:427–432
Hommais F, Gouriou S, Amorin C, Bui H, Rahimy MC, Picard B, Denamur E (2003) The FimH A27V mutation is pathoadaptive for urovirulence in Escherichia coli B2 phylogenetic group isolates. Infect Immun 71:3619–3622
Klemm P, Christiansen G (1987) Three fim genes required for the regulation of length and mediation of adhesion of Escherichia coli type 1 fimbriae. Mol Gen Genet 208:439–445
Lancaster A, Nelson MP, Single RM, Meyer D, Thomson G (2003) PyPop:a software framework for population genomics:analyzing large-scale multi-locus genotype data. In: Altman RB, et al. (eds) Pacific Symposium on Biocomputing 8. World Scientific, Singapore, pp 514–525
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Orr MR, Smith TB (1998) Ecology and speciation. Trends Ecol Evol 13:502–506
Pielou EC (1969) An introduction to mathematical ecology. Wiley-Interscience, New York, pp 203–233
Pulliam HR (1988) Sources, sinks, and population regulation. Am Nat 132:652–661
Salamon H, Klitz W, Easteal S, Gao X, Erlich HA, Fernandez–Viña M, Trachtenberg EA, McWeeney SK, Nelson MP, Thomson G (1999) Evolution of HLA class II molecules: allelic and amino acid site variability across populations. Genetics 152:393–400
Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Möllby R, Winberg J, Normark S (1994) The Gal(alpha 1-4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc Natl Acad Sci USA 91:11889–11893
Simpson EH (1949) Measurement of diversity. Nature 163:688
Slatkin M (1994) An exact test for neutrality based on the Ewens sampling distribution. Genet Res 64:71–74
Slatkin M (1996) A correction to the exact test based on the Ewens sampling distribution. Genet Res 68:259–260
Sokurenko EV, Chesnokova V, Doyle RJ, Hasty DL (1997) Diversity of the Escherichia coli type 1 fimbrial lectin. Differential binding to mannosides and uroepithelial cells. J Biol Chem 272:17880–17886
Sokurenko EV, Courtney HS, Maslow J, Siitonen A, Hasty DL (1995) Quantitative differences in adhesiveness of type 1 fimbriated Escherichia coli due to structural differences in fimH genes. J Bacteriol 177:3680–3686
Sokurenko EV, Chesnokova V, Dykhuzien DE, Ofek I, Wu X-R, Krogfelt KA, Struve C, Schembri MA, Hasty DL (1998) Pathogenic adaptation of Escherichia coli by natural variation of the FimH adhesin. Proc Natl Acad Sci USA 95:8922–8926
Sokurenko EV, Feldgarden M, Trintchina E, Weissman SJ, Avagyan S, Chattopadhyay S, Johnson JR, Dykhuizen DE (2004) Selection footprint in the FimH adhesin shows pathoadaptive niche differentiation in Escherichia coli. Mol Biol Evol 21:1373–1383
Sokurenko EV, Gomulkiewicz R, Dykhuizen DE (2006) Source–sink dynamics of virulence evolution. Nat Rev Microbiol 4:548–555
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphisms. Genetics 123:585–595
Watterson GA (1978) The homozygosity test of neutrality. Genetics 88:405–417
Weissman SJ, Chattopadhyay S, Aprikian P, Obata-Yasuoka M, Yarova-Yarovaya Y, Stapleton A, Ba-Thein W, Dykhuizen D, Johnson JR, Sokurenko EV (2006) Clonal analysis reveals high rate of structural mutations in fimbrial adhesins of extraintestinal pathogenic Escherichia coli. Mol Microbiol 59:975–988
Welch RA, Burland V, Plunkett III G, Redford P, Roesch P, Rasko D, Buckles EL, Llou S-R, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HLT, Donnenberg MS, Blattner FR (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA 99:17020–17024
White GC, Bennetts RE (1996) Analysis of frequency count data using the negative binomial distribution. Ecology 77:2549–2557
Yang Z (1997) PAML:a program package for phylogenetic analysis by maximum likelihood. Comput Appl BioSci 13:555–556
Acknowledgments
This study is supported by NIH grants GM60731 and DK053369.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Margaret Riley]
Rights and permissions
About this article
Cite this article
Chattopadhyay, S., Feldgarden, M., Weissman, S.J. et al. Haplotype Diversity in “Source-Sink” Dynamics of Escherichia coli Urovirulence. J Mol Evol 64, 204–214 (2007). https://doi.org/10.1007/s00239-006-0063-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0063-5