
Abstract
Members of the protein phosphatase 2C (PP2C) superfamily are Mg2+/Mn2+-dependent serine/threonine phosphatases, which are essential for regulation of cell cycle and stress signaling pathways in cells. In this study, a comprehensive genomic analysis of all available metazoan PP2C sequences was conducted. The phylogeny of PP2C was reconstructed, revealing the existence of 15 vertebrate families which arose following a series of gene duplication events. Relative dating of these duplications showed that they occurred in two active periods: before the divergence of bilaterians and before vertebrate diversification. PP2C families which duplicated during the first period take part in different signaling pathways, whereas PP2C families which diverged in the second period display tissue expression differences yet participate in similar signaling pathways. These differences were found to involve variation of expression in tissues which show higher complexity in vertebrates, such as skeletal muscle and the nervous system. Further analysis was performed with the aim of identifying the functional domains of PP2C. The conservation pattern across the entire PP2C superfamily revealed an extensive domain of more than 50 amino acids which is highly conserved throughout all PP2C members. Several insertion or deletion events were found which may have led to the specialization of each PP2C family.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reversible protein phosphorylation is a major regulatory mechanism of cellular functions such as stress-activated signal transduction, mitogenic signal transduction, and cell cycle control (e.g., Sun and Tonks 1994; Hanada et al. 2001). Protein kinases have been in the spotlight for several decades (Burnett and Kennedy 1954; Bolen 1995; Shokat 1995; Yeh and Pellegrini 1999; Schlessinger 2000; Cowan and Storey 2003), leading to the recent development of drug therapy using kinase inhibitors (reviewed by Krause and Van Etten 2005). Yet far less focus has been given to the kinases counterparts in cell regulation, the protein phosphatases (PPs). It is becoming clear that the interplay between kinases and phosphatases is quite complex. Thus, in order to fully understand the process of phosphorylation, it is imperative to focus research on phosphatases as well as on kinases.
Protein serine/threonine phosphatases are divided into four structurally distinct superfamilies (Cohen 1989; Shenolikar 1994; Barford, Jia, and Tonks 1995; Wera and Hemmings 1995): PP1, PP2A, PP2B, and PP2C. The PP2C superfamily of phosphatases (also referred to as PPM) is defined by distinct amino acid sequence and three-dimensional (3D) structure (Tamura et al. 1989; Mann et al. 1992; Wenk et al. 1992). The PP2C superfamily does not seem to be evolutionarily related to PP1, PP2A, and PP2B, which are all multisubunit enzymes. This study focuses on the PP2C superfamily as an embodiment of the evolutionary diversity of protein phosphatases.
PP2C Functions
PP2C is a monomeric enzyme which displays broad substrate specificity. Distinguishing characteristics of PP2C are its (i) absolute requirement for divalent cations, mainly Mg+2 or Mn+2; (ii) distinctive structural features; and (iii) insensitivity to inhibition by okadaic acid (Barford et al. 1995; Wera and Hemmings 1995). At least 15 distinct PP2C human paralogues have been found in mammalian cells (Table 1). All of these PP2Cs have Mg+2- and/or Mn+2-dependent phosphatase activity against artificial substrates in vitro (Komaki et al. 2003).
The majority of PP2Cs are involved in regulation of stress activated protein kinase (SAPK) cascades which relay signals in response to external stimuli (Meskiene et al. 2003). These cascades are a subfamily of the mitogen-activated protein kinase (MAPK) cascades. Different PP2Cs negatively regulate SAPK pathways at different levels. For instance, PP2Cβ inhibits the TAK1 pathway (Hanada et al. 2001; Li et al. 2003) and is involved in the NF-κB pathway (Prajapati et al. 2004). PP2Cα inactivates the p38 pathway and the c-Jun amino-terminal kinase (JNK) pathway (Takekawa et al. 1998) and is additionally involved in the Wnt signaling pathway (Strovel et al. 2000).
Recent studies have demonstrated that the PP2C superfamily is also associated with eukaryotic cell cycle processes, which are controlled by the ordered activation and inactivation of cyclin-dependent protein kinases (CDKs). Reversible protein phosphorylation is one of the mechanisms through which extracellular and intracellular signals regulate CDKs (Cheng et al. 1999, 2000). Additionally, we have previously reported that overexpression of PP2Cα activates the expression of the tumor suppressor gene TP53/p53, which leads to G2/M cell cycle arrest and apoptosis (Ofek et al. 2003). Thus, the PP2C superfamily appears to be directly involved in several cell regulation and cell signaling processes. In fact, many PP2C members have been reported to inactivate CDK and MAPK family kinases by dephosphorylating a conserved threonine residue on the so-called T-loop of these kinases (Marley et al. 1996; Cheng et al. 1999; Takekawa et al. 2000), implicating that the PP2C superfamily may be general T-loop phosphatases.
Phylogenetic Study of PP2C
An evolutionary comparison of kinomes across species (Manning et al. 2002) has demonstrated the value of a phylogenetic study of kinases. This enabled mapping kinases specific to each lineage, as well as delineating the pathways involving kinases shared throughout various evolutionary lineages. Similar to families of protein kinases, the multiplicity of PP2C proteins suggests a broad functional diversity of these proteins. Thus, the aim of this study is to conduct a comprehensive genomic evolutionary analysis of all known members of the PP2C superfamily in Metazoa. This is the first analysis of its kind undertaken in the study of the PP2C superfamily and, as such, promises to be highly informative in characterizing protein isoform diversification. Consequently, we performed an extensive search of all genomic databases for PP2C genes. We then conducted a phylogenetic analysis of all PP2C members found, with the aim of assigning paralogy and orthology relations to each sequence found. These assignments enabled us to predict functions of previously unidentified genes in the superfamily, as well as explore the differences between the families within the PP2C superfamily and estimate the relative dates of the diversification events. We thereby explore the breadth of PP2C functional conservation throughout the metazoan kingdom.
Methods
Search for PP2C Members in Metazoa
Sequences were retrieved from the following databases: GenBank (http://www.ncbi.nlm.nih.gov; Benson et al. 2005), ENSEMBL (http://www.ensembl.org; Hubbard et al. 2005), FLYBASE (http://www.fruitfly.org; Stapleton et al. 2002), and WORMBASE release WS130 (http://www.wormbase.org; Harris et al. 2004). Initially, all fully sequenced non-plant eukaryote genomes (Homo sapiens, Pan troglodytes, Mus musculus, Rattus norvegicus, Canis familiaris, Bos taurus, Monodelphis domestica, Gallus gallus, Xenopus tropicalis, Danio rerio, Takifugu rubripes, Tetraodon nigroviridis, Drosophila melanogaster, Anopheles gambiae, Apis mellifera, Caenorhabditis elegans, Saccharomyces cerevisiae) were screened for PP2C sequences using BLAT (Kent 2002), TBLASTN (Altschul et al. 1990), and the Orthologue Prediction section of ENSEMBL. Sequences uncovered in these stages were filtered according to two criteria: (i) due to an unreliable alignment containing excess gaps, all yeast sequences were discarded; and (ii) orthologous sequences for which there was a significant deviation from the PP2C signature defined in PROSITE (Bairoch and Bucher 1994; entry PS01032) were discarded. Accession numbers of all sequences used in the study are available as supplementary material.
Sequence Alignment and Phylogenetic Reconstruction
Multiple sequence alignment (MSA) was performed using the MUSCLE program version 3.52 (Edgar 2004). Maximum likelihood-based phylogenetic reconstruction was performed with the PhyML program (Guindon et al. 2005) using among-site rate variation with four discrete rate categories and the JTT model of sequence evolution. Node supports were determined by performing 100 bootstrap replicates (Felsenstein 1985). Bootstrap values higher than 70% were considered significant.
Due to the extended evolutionary time spanned by the sequences in this analysis and the low similarity between different paralogues, the resulting alignment includes regions which are difficult to align as well as many gaps. This is a general problem when analyzing superfamily genes, and one must verify that the inferred phylogeny does not depend on those parts of the alignment which are uncertain. In order to test the robustness of the results to the alignment’s validity, we ran the Gblocks program (Castresana 2000) on the alignment, with the aim of removing poorly aligned regions and then performed the phylogenetic analysis on the resulting positions. The new tree was found to be essentially identical to the tree which was obtained based on the full alignment. All minor differences between the trees were supported by low bootstrap values in the new tree. The settings used to run Gblocks, together with the resulting reduced alignment and resulting phylogenetic tree, are given in the supplementary material.
A specific sequence was considered part of a group if it belonged to a monophyletic clade in the tree. For this purpose, it was assumed that the root of the tree is on one of the branches leading to one of the more anciently derived groups (as defined in the Results section).
Site-Specific Evolutionary Rate Analysis
The conservation pattern of the PP2C superfamily was analyzed by estimating site-specific evolutionary rates, using the Bayesian approach of the Consurf server (Mayrose et al. 2004; Landau et al. 2005). The analysis was conducted using the reconstructed PhyML tree. Site-specific positive selection was analyzed using the Selecton server (Doron-Faigenboim et al. 2005), using the Bayesian method (Yang et al. 2000). Once again, the reconstructed PhyML tree was given as input.
Results
The search for PP2C members in the human genome led to the identification of 15 members (Table 1). Each of these members was found throughout the vast majority of the vertebrate genomes searched, with the exception of a few orthologues not found. It is unclear whether these exceptions stemmed from incomplete sequencing or from gene losses in these organisms. All the 15 groups of orthologues clustered as monophyletic groups in the phylogenetic tree reconstructed (Fig. 1), providing firm phylogenetic support for the hypothesis whereby the PP2C superfamily arose following a series of duplication events and for the classical classification of the PP2C family members. Furthermore, the phylogenetic tree enabled assigning annotation to previously unidentified genes according to their location on the phylogenetic tree.

Maximum likelihood phylogenetic tree of PP2C, with the TA group used as an outgroup. For brevity, the tree presented here shows a collapsed version of the full tree, where all clades representing vertebrate orthologues were collapsed, as were all clades of orthologous insects (collapsed clades are shown as black triangles). The lengths of the branches leading to the collapsed groups are the lengths of the branches leading to the ancestral groups in the original tree. Major PP2C groups are shaded in color for clarity. The branch colored yellow represents a putative gene duplication event with subsequent gene loss in vertebrates in one of the duplicants. The scale bar represents one branch length unit, which is equivalent to an average of one substitution per site. A full tree in Newick format is available in the supplementary material.
Two Types of PP2C
Mammalian PP2Cs have been previously classified into two subgroups according to differences in amino acid sequence motifs. Group 2 consists of PP2Cη, PP2Cζ, and PPM1H, while group 1 includes all other PP2Cs (Komaki et al. 2003). TA-PP2C, discovered only a year later (Mao et al. 2004), was not classified as belonging to any of the two groups . Groups 1 and 2 differ in their PP2C signature, as well as in the other sequential motifs found to characterize the PP2C superfamily (Komaki et al. 2003). All residues forming the catalytic domain (Das et al. 1996) are part of the PP2C signature and these additional motifs.
The phylogenetic reconstruction of the PP2C family (Fig. 1) shows that the three families belonging to group 2 (PP2Cη, PP2Cζ, and PPM1H) form a monophyletic clade (99% bootstrap), supporting the previous finding whereby group 2 is characterized by a unique sequence (Komaki et al. 2003). Since the differences between group 1 and group 2 are displayed in residues which surround the catalytic site, this may hint at a functional divergence of the PP2C superfamily into two functionally distinct groups.
Mapping of PP2C Duplications
Nine different clades in the tree comprise members in protostomes. These include sequences belonging to monophyletic PP2C groups (TA-PP2C, ILKAP, PP2Cγ, Wip1, and PP2Cε), as well as sequences assumed to have been derived from ancestral forms of PP2C prior to their duplication. For example, a worm sequence which forms an outgroup of the PDP1 and PDP2 vertebrate clades is assumed to be derived from the ancestral sequence of PDP1 and PDP2 prior to their duplication and will be referred to as the PDP1|PDP2 ancestrally derived sequence. Similarly, there is a protostome ancestrally derived sequence of POPX1|FEM2, PP2Cα|PP2Cβ, group 2 (PP2Cη|PP2Cζ|PPM1H), and PDP1|PDP2. Consequently, it appears that much of the PP2C diversification occurred before bilaterian diversification. On the other hand, the nine paralogous groups, PP2Cα, PP2Cβ, POPX1, FEM2, PP2Cζ, PPM1H, PPM1K, PDP1, and PDP2, are only present in vertebrates. Therefore, the mapping of the emergence of each one of the PP2C groups during evolution is possible, as depicted in Fig. 2. Nine groups emerged prior to the divergence of protostomes, while another nine were created by duplications prior to the divergence of vertebrates.

The two active periods of gene diversification by duplication are marked by arrows in the currently accepted species tree (Halanych 2004). The rectangle represents the putative emergence of the groups: PP2Cα, PP2Cβ, PPM1K, FEM2, POPX1, PP2Cζ, PPM1H, PDP1, and PDP2. The diamond represents the latest possible dating of the emergence of the groups: Wip1, ILKAP, PP2Cγ, TA-PP2C, PP2Cε, PP2Cα|β, PDP1|PDP2, group 2, and FEM2|POPX1.
More precise mapping and relative dating of the different gene duplication events proved to be more difficult. A majority of the more ancient duplications were supported by very low bootstrap values on the tree (Fig. 1). Thus, it is currently impossible to map which members were created by each ancient duplication event, and to determine the order of duplications. This is further aggravated by lack of available genomes, specifically in nonvertebrate chordates and poriferans. However, the formation of four more recent duplications was supported by very high bootstrap values. These duplications (PP2Cα versus PP2Cβ, PDP1 versus PDP2, PP2Cζ versus PPM1H, and POPX1 versus FEM2) occurred before vertebrate diversification and are supported by a single form in nonvertebrates (sea urchin and/or insects). For example, the clades of PDP1 and PDP2 do not include any sea urchin sequences, yet a sea urchin sequence forms an outgroup. An additional duplication, between PP2Cη and the PP2Cζ|PPM1H ancestor, is more difficult to map precisely. According to the tree, the duplication occurred after the speciation of insects and before the speciation of vertebrates. However, it is unclear whether this duplication occurred before or after the speciation of sea urchin, due to the location of the two sea urchin sequences (Fig. 1; denoted Group 2 S. purpuratus 1 and 2).
Protostome-Specific Duplications
The phylogenetic tree supports an ancient duplication event in the ancestral PP2Cγ (Fig. 1; marked in yellow) where the gene was apparently lost in the lineage leading to vertebrates yet remained in protostomes. This novel paralogue further underwent another duplication in insects. Yet another insect-specific duplication is evident, as is apparent by the existence of both α|β D. melanogaster 2 and α|β D. melanogaster 1 (see Fig. 1). In both these insect-specific duplications, no sequence is available from A. gambiae, which may indicate either a loss in this lineage or missing data in the A. gambiae genome. Little is known of these expansions in insects, yet they are supported by EST evidence in D. melanogaster (UCSC genome browser [Kent et al. 2002]), ruling out the hypothesis whereby these sequences represent pseudogenes.
Mapping Functional Regions in PP2C
We performed a comprehensive analysis of the pattern of amino acid conservation throughout the MSA of the PP2C superfamily. This analysis took into account the phylogenetic tree, thus enabling more precise inference of conservation of amino acid sites (Pupko et al. 2002). Two crystal structures of PP2C members exist in the Protein Data Bank (Berman et al. 2000)—one of the human PP2Cα (Das et al. 1996) and one of the Mycobacterium tuberculosis PP2C (Pullen et al. 2004). The conservation pattern obtained for the PP2C superfamily was mapped onto the Van-der-Waals surface of PP2Cα, since it is the only metazoan PP2C with a known 3D structure. A clear pattern of high conservation is apparent throughout the N-terminus of the protein, while the C-terminus of the protein is highly variable. The variability of the C-terminus is expected, since the C-terminus of the PP2Cα is unique to this family and may serve as a substrate recognition domain in the cleft that is created between it and the catalytic domain (Das et al. 1996). However, the relatively high conservation pattern of the N-terminus is more surprising. On the one hand, the results reinforce the previous knowledge of functionally important sites in PP2C, showing that the nine catalytic residues found in PP2Cα (Das et al. 1996) are indeed highly conserved. More surprisingly, over 50 additional sites appeared to be highly conserved. While some of these sites cluster around the catalytic site in the globular N-terminus, others form the cleft between the N-terminus and the C-terminus and form part of the bulk above the catalytic region (according to the orientation in Fig. 3). The high conservation of the N-terminal part of the cleft suggests that there may be a shared mechanism of this cleft throughout all the PP2C families.

The conservation pattern of the PP2C superfamily as inferred by Consurf (Landau et al. 2005). Conservation scores are color-coded onto the Van der Waals surface of PP2Cα, where maroon corresponds to maximal conservation, white corresponds to average conservation, and turquoise corresponds to maximal variability. The Mg2+ ions and associated water molecules are shown in yellow, and the nine previously identified catalytic sites (Das et al. 1996) are shown in red. These nine sites also attained the highest level of conservation in the analysis. Arrows show the globular N-terminus region and the C-terminus tail.
In order to study the differences between the PP2C families, pairs of PP2C families were analyzed for site-specific positive selection using the Selecton server (Doron-Faigenboim et al. 2005). The underlying assumption was that following the gene duplication events which created the two families, both genes underwent a specialization process. Such a process may have led to a rapid fixation of mutations due to positive selection forces. Thus, all pairs of PP2C families which most recently diverged (PP2Cα-PP2Cβ, PDP1-PDP2, FEM2-POPX1, PPM1H-PP2Cζ) were analyzed. The analysis of pairs of sequences, as opposed to an analysis of the entire superfamily, enables a more reliable MSA at the codon level. However, in all of these families no site-specific or global positive selection was observed. This may be due to the fact that purifying selection within each of the families obscures the footprint of positive selection which the families underwent, and due to the small species sampling.
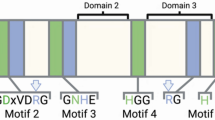
Additional evidence for gene specialization may be obtained by analyzing insertion and deletion events in the different gene families. To this end, the MSA was utilized as a rough indicator of insertion and deletion events (using visual inspection). A schematic drawing was created depicting PP2C domains common to all groups, as well as those unique to specific groups (Fig. 4). Only such blocks which are well defined and clearly observed when viewing the alignment were depicted. Furthermore, these blocks were flanked by anchors of regions which are conserved throughout the entire alignment. This analysis suggests that the evolution of the PP2C family included several significant insertion or deletion events which may have led to the specialization of the duplicants.

A schematic drawing of the PP2C superfamily alignment. Bars represent blocks which are unique to specific groups. Gray rectangles represent the rest of the alignment, including all regions which are common to all PP2C proteins. Coordinates of the blocks (according to the MSA) appear underneath each block. The MSA is available as part of the supplementary material.
Discussion
In this study, we present an analysis of PP2C evolution. The reconstructed phylogeny displays the relationship between PP2C paralogues and orthologues throughout Metazoa, revealing the existence of at least 15 PP2C groups which were created via gene duplication. Analysis of the PP2C superfamily suggests that two waves of duplications were responsible for the creation of the majority of the PP2C members. The first wave of duplications presumably led to the formation of functionally different groups which specialized in different catalytic processes. Our analysis suggests that this wave took place before the divergence of bilaterians. Presumably, these ancient duplications occurred successively in a short time frame (represented by short branches between the different groups; see Fig. 1), rendering it difficult to determine the order of the duplication events (as is evident from the low bootstrap support on these branches). The second wave of duplications presumably led to the formation of tissue- specific groups, and most likely took place at the beginning of vertebrate evolution. More precise timing of these duplication events is still beyond reach due to the lack of sequence data in poriferans.
Extensive gene duplication events during early chordate evolution have been previously reported (e.g., Miyata and Suga 2001; McLysaght et al. 2002). These extensive events may have been the result of a whole-genome duplication or a series of partial chromosomal block duplications. These duplications, which took place in early vertebrate evolution, are thought to account for the existence of the four HOX gene clusters (Larhammar et al. 2002; Prince 2002), as well as for the four different MHC gene clusters (Abi-Rached et al. 2002). Numerous studies have found protein families with a pattern of evolution similar to the pattern of PP2C found here (e.g., Miyata and Suga 2001; Wakeham et al. 2005). Many of these families include kinase and phosphatase families involved in cell signaling. Furthermore, a comparative study comparing kinome catalogs throughout different species (Manning et al. 2002) revealed that the creation of functionally distinct kinase families occurred during early metazoan evolution. Thus, we postulate that the evolution of signal transduction occurred in two major active periods. The first, occurring before metazoan radiation, may have been driven by the increase in complexity of multicellular organisms. This required more sophisticated signaling between and within cells. Furthermore, the more complex developmental mechanisms also required a more elaborate network of signaling proteins. This suggests that the more anciently derived PP2Cs evolved following a requirement for new signaling pathways. For example, the more anciently derived Wip1 and ILKAP display this pattern: whereas Wip1 evolved as part of the cell cycle pathway, ILKAP evolved to participate in cell adhesion and growth factor signaling.
The second active period, before vertebrate diversification, occurred concomitantly with the development of tissues such as skeletal muscle and the nervous system. Indeed, the transition between nonvertebrates and vertebrates is believed to be one of the major leaps in complexity during evolution, involving the evolution of cells such as the neural crest, the brain, and the spinal cord (Gilbert 2001). Insights into this phenomenon are evident throughout all PP2C groups found to have diverged in this second active period. All PP2C vertebrate-specific duplications characterized in this study show specialized tissue-specific expression patterns. In fact, in each of these duplications, one duplicant is uniquely expressed in either skeletal muscle or nervous system tissue. Whereas PP2Cα and FEM2 are ubiquitous in their duplication partners PP2Cβ and POPX1 are tissue specific. PP2Cβ splice variants were shown to display skeletal muscle- and heart-specific tissue expression (Marley et al. 1998; Seroussi et al. 2001). Similarly, POPX1 displays brain-specific expression (Koh et al. 2002). PP2Cζ is displayed in the testicular germ cells (Kashiwaba et al. 2003), while PPM1H is expressed in the brain and is involved in neuronal inhibitory pathways (Labes et al. 1998). Finally, PDP1 and PDP2 both catalyze the same reaction but differ in tissue distribution. PDP1 is highly expressed in skeletal muscle, whereas PDP2 is expressed in liver (Huang et al. 1998).
The evolutionary conservation pattern of PP2C suggests that the highly conserved catalytic domain and the surrounding core are shared by all PP2C members. Of specific interest are those residues which have not been previously identified as critical to the enzymatic function of the protein. Since these sites are abundant on the protein surface, they may represent novel protein or ligand binding sites. As the PP2C superfamily plays an important role in delicate signals relayed across the cell, it is likely that the different PP2C proteins are further bound by tight regulation. Thus, the novel conserved sites found may be the key to understanding these regulatory mechanisms.
Aside from the shared conserved domain, several PP2C families have a unique appendage which may be the specificity determinant of this family. For instance, in the C-terminal region, PP2Cα and PP2Cβ share a unique tail (Fig. 4). These differences may be responsible for the fact that the two paralogues differ in their substrate binding abilities. Further investigation of the differences between the families could also focus on the gene-regulation level, for instance, by comparing the promoters and nontranslated regions of the different paralogues. We have previously reported that the nontranslated regions of PP2Cβ are highly conserved throughout orthologous genes (Seroussi et al. 2001), indicating a significant regulatory role for these regions. Furthermore, different PP2Cβ transcripts were found to differ by alternative splicing and alternative promoters (Ohnishi et al. 1999). It thus seems that the diversification which gave rise to new PP2C families continued with the creation of variants which differ at the transcriptional level. Pinpointing the precise functional differences among and within the PP2C families remains a challenge which may play a pivotal role in our understanding of complex signal networks in cells.
References
Abi-Rached L, Gilles A, Shiina T, Pontarotti P, Inoko H (2002) Evidence of en bloc duplication in vertebrate genomes. Nat Genet 31:100–105
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bairoch A, Bucher P (1994) PROSITE: recent developments. Nucleic Acids Res 22:3583–3589
Barford D, Jia Z, Tonks NK (1995) Protein tyrosine phosphatases take off. Nat Struct Biol 2:1043–1053
Behal RH, Buxton DB, Robertson JG, Olson MS (1993) Regulation of the pyruvate dehydrogenase multienzyme complex. Annu Rev Nutr 13:497–520
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2004) GenBank: update. Nucleic Acids Res 32:D23–D26
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2005) GenBank. Nucleic Acids Res 33:D34–D38
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Bolen JB, (1995) Protein tyrosine kinases in the initiation of antigen receptor signaling. Curr Opin Immunol 7:306–311
Burnett G, Kennedy EP (1954) The enzymatic phosphorylation of proteins. J Biol Chem 211:969–980
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Cheng A, Ross KE, Kaldis P, Solomon MJ (1999) Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev 13:2946–2957
Cheng A, Kaldis P, Solomon MJ (2000) Dephosphorylation of human cyclin-dependent kinases by protein phosphatase type 2C alpha and beta 2 isoforms. J Biol Chem 275:34744–34749
Cohen P (1989) The structure and regulation of protein phosphatases. Annu Rev Biochem 58:453–508
Cowan KJ, Storey KB (2003) Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol 206:1107–1115
Das AK, Helps NR, Cohen PT, Barford D (1996) Crystal structure of the protein serine/threonine phosphatse 2C at 2.0 A resolution. EMBO J 15:6798–6809
Doron-Faigenboim A, Stern A, Mayrose I, Bacharach E, Pupko T (2005) Selecton: a server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 21:2101–2103
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Felsenstein J (1985) Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791
Gaits F, Shiozaki K, Russell P (1997) Protein phosphatase 2C acts independently of stress-activated kinase cascade to regulate the stress response in fission yeast. J Biol Chem 272:17873–17879
Gilbert SF (2001) Developmental mechanisms of evolutionary change. In: Developmental biology, 6th ed. Sinauer Associates, Sunderland, MA, pp 688–689
Guindon S, Lethiec F, Duroux P, Gascuel O (2005) PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33:W557–W559
Halanych KM (2004) The new view of animal phylogeny. Annu Rev Ecol Evol Syst 35:229–256
Hanada M, Ninomiya-Tsuji J, Komaki K, Ohnishi M, Katsura K, Kanamaru R, Matsumoto K, Tamura S (2001) Regulation of the TAK1 signaling pathway by protein phosphatase 2C. J Biol Chem 276:5753–5759
Harris TW, Chen N, Cunningham F, Tello-Ruiz M, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Bradnam K, Chan J, Chen CK, Chen WJ, Davis P, Kenny E, Kishore R, Lawson D, Lee R, Muller HM, Nakamura C, Ozersky P, Petcherski A, Rogers A, Sabo A, Schwarz EM, Van Auken K, Wang Q, Durbin R, Spieth J, Sternberg PW, Stein LD (2004) WormBase: a multi-species resource for nematode biology and genomics. Nucleic Acids Res 32:D411–D417
Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM (1998) Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem 273:17680–17688
Hubbard T, Andrews D, Caccamo M, Cameron G, Chen Y, Clamp M, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T, Down T, Durbin R, Fernandez-Suarez XM, Gilbert J, Hammond M, Herrero J, Hotz H, Howe K, Iyer V, Jekosch K, Kahari A, Kasprzyk A, Keefe D, Keenan S, Kokocinsci F, London D, Longden I, McVicker G, Melsopp C, Meidl P, Potter S, Proctor G, Rae M, Rios D, Schuster M, Searle S, Severin J, Slater G, Smedley D, Smith J, Spooner W, Stabenau A, Stalker J, Storey R, Trevanion S, Ureta-Vidal A, Vogel J, White S, Woodwark C, Birney E (2005) Ensembl 2005. Nucleic Acids Res 33:D447–D453
Kashiwaba M, Katsura K, Ohnishi M, Sasaki M, Tanaka H, Nishimune Y, Kobayashi T, Tamura S (2003) A novel protein phosphatase 2C family member (PP2Czeta) is able to associate with ubiquitin conjugating enzyme 9. FEBS Lett 538:197–202
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12:996–1006
Koh CG, Tan EJ, Manser E, Lim L (2002) The p21-activated kinase PAK is negatively regulated by POPX1 and POPX2, a pair of serine/threonine phosphatases of the PP2C family. Curr Biol 12:317–321
Komaki K, Katsura K, Ohnishi M, Guang Li M, Sasaki M, Watanabe M, Kobayashi T, Tamura S (2003) Molecular cloning of PP2Ceta, a novel member of the protein phosphatase 2C family. Biochim Biophys Acta 1630:130–137
Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353:172–187
Labes M, Roder J, Roach A (1998) A novel phosphatase regulating neurite extension on CNS inhibitors. Mol Cell Neurosci 12:29–47
Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-TalN (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33:W299–W302
Larhammar D, Lundin LG, Hallbook F (2002) The human Hox-bearing chromosome regions did arise by block or chromosome (or even genome) duplications. Genome Res 12:1910–1920
Lawson JE, Park SH, Mattison AR, Yan J, Reed LJ (1997) Cloning, expression, and properties of the regulatory subunit of bovine pyruvate dehydrogenase phosphatase. J Biol Chem 272:31625–31629
Leung-Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE (2001) Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J 20:2160–2170
Li MG, Katsura K, Nomiyama H, Komaki K, Ninomiya-Tsuji J, Matsumoto K, Kobayashi T, Tamura S (2003) Regulation of the interleukin-1-induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cepsilon). J Biol Chem 278:12013–12021
Mann DJ, Campbell DG, McGowan CH, Cohen PT (1992) Mammalian protein serine/threonine phosphatase 2C: cDNA cloning and comparative analysis of amino acid sequences. Biochim Biophys Acta 1130:100–104
Manning G, Plowman GD, Hunter T, Sudarsanam S (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci 27:514–520
Mao M, Biery MC, Kobayashi SV, Ward T, Schimmack G, Burchard J, Schelter JM, Dai H, He YD, Linsley PS (2004) T lymphocyte activation gene identification by coregulated expression on DNA microarrays. Genomics 83:989–999
Marley AE, Sullivan JE, Carling D, Abbott WM, Smith GJ, Taylor IW, Carey F, Beri RK (1996) Biochemical characterization and deletion analysis of recombinant human protein phosphatase 2C alpha. Biochem J 320(Pt 3):801–806
Marley AE, Kline A, Crabtree G, Sullivan JE, Beri RK (1998) The cloning expression and tissue distribution of human PP2Cbeta. FEBS Lett 431:121–124
Mayrose I, Graur D, Ben-Tal N, Pupko T (2004) Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol 21:1781–1791
McLysaght A, Hokamp K, Wolfe KH (2002) Extensive genomic duplication during early chordate evolution. Nat Genet 31:200–204
Meskiene I, Baudouin E, Schweighofer A, Liwosz A, Jonak C, Rodriguez PL, Jelinek H, Hirt H (2003) Stress-induced protein phosphatase 2C is a negative regulator of a mitogen-activated protein kinase. J Biol Chem 278:18945–18952
Miyata T, Suga H (2001) Divergence pattern of animal gene families and relationship with the Cambrian explosion. Bioessays 23:1018–1027
Murray MV, Kobayashi R, Krainer AR (1999) The type 2C Ser/Thr phosphatase PP2Cgamma is a pre-mRNA splicing factor. Genes Dev 13:87–97
Ofek P, Ben-Meir D, Kariv-Inbal Z, Oren M, Lavi S (2003) Cell cycle regulation and p53 activation by protein phosphatase 2C alpha. J Biol Chem 278:14299–14305
Ohnishi M, Chida N, Kobayashi T, Wang H, Ikeda S, Hanada M, Yanagawa Y, Katsura K, Hiraga A, Tamura S (1999) Alternative promoters direct tissue-specific expression of the mouse protein phosphatase 2Cbeta gene. Eur J Biochem 263:736–745
Prajapati S, Verma U, Yamamoto Y, Kwak YT, Gaynor RB (2004) Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem 279:1739–1746
Prince V (2002) The Hox paradox: more complex(es) than imagined. Dev Biol 249:1–15
Pullen KE, Ng HL, Sung PY, Good MC, Smith SM, Alber T (2004) An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C-family Ser/Thr protein phosphatase. Structure 12:1947–1954
Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N (2002) Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18 (Suppl 1):S71–S77
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Seroussi E, Shani N, Ben-Meir D, Chajut A, Divinski I, Faier S, Gery S, Karby S, Kariv-Inbal Z, Sella O, Smorodinsky NI, Lavi S (2001) Uniquely conserved non-translated regions are involved in generation of the two major transcripts of protein phosphatase 2Cbeta. J Mol Biol 312:439–451
Shenolikar S (1994) Protein serine/threonine phosphatases—new avenues for cell regulation. Annu Rev Cell Biol 10:56–86
Shokat KM (1995) Tyrosine kinases: modular signaling enzymes with tunable specificities. Chem Biol 2:509–514
Stapleton M, Carlson J, Brokstein P, Yu C, Champe M, George R, Guarin H, Kronmiller B, Pacleb J, Park S, Wan K, Rubin GM, Celniker SE (2002) A Drosophila full-length cDNA resource. Genome Biol 3:RESEARCH0080
Strovel ET, Wu D, Sussman DJ (2000) Protein phosphatase 2Calpha dephosphorylates axin and activates LEF-1-dependent transcription. J Biol Chem 275:2399–2403
Sun H, Tonks NK (1994) The coordinated action of protein tyrosine phosphatases and kinases in cell signaling. Trends Biochem Sci 19:480–485
Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K (2000) p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 19:6517–6526
Takekawa M, Maeda T, Saito H (1998) Protein phosphatase 2Calpha inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J 17:4744–4752
Tamura S, Lynch KR, Larner J, Fox J, Yasui A, Kikuchi K, Suzuki Y, Tsuiki S (1989) Molecular cloning of rat type 2C (IA) protein phosphatase mRNA. Proc Natl Acad Sci USA 86:1796–1800
Terasawa T, Kobayashi T, Murakami T, Ohnishi M, Kato S, Tanaka O, Kondo H, Yamamoto H, Takeuchi T, Tamura S (1993) Molecular cloning of a novel isotype of Mg(2+)-dependent protein phosphatase beta (type 2C beta) enriched in brain and heart. Arch Biochem Biophys 307:342–349
Travis SM, Welsh MJ (1997) PP2C gamma: a human protein phosphatase with a unique acidic domain. FEBS Lett 412:415–419
Wakeham DE, Abi-Rached L, Towler MC, Wilbur JD, Parham P, Brodsky FM (2005) Clathrin heavy and light chain isoforms originated by independent mechanisms of gene duplication during chordate evolution. Proc Natl Acad Sci USA 102:7209–7214
Wenk J, Trompeter HI, Pettrich KG, Cohen PT, Campbell DG, Mieskes G (1992) Molecular cloning and primary structure of a protein phosphatase 2C isoform. FEBS Lett 297:135–138
Wera S, Hemmings BA (1995) Serine/threonine protein phosphatases. Biochem J 311(Pt 1):17–29
Yamaguchi H, Minopoli G, Demidov ON, Chatterjee DK, Anderson CW, Durell SR, Appella E (2005) Substrate specificity of the human protein phosphatase 2Cdelta, Wip1. Biochemistry 44:5285–5294
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yeh TC, Pellegrini S (1999) The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell Mol Life Sci 55:1523–1534
Acknowledgments
We thank Dorothee Huchon for her insightful comments. A.S. is a fellow of the Edmond J. Safra Program in Bioinformatics at Tel Aviv University. S.L was supported by the Linda Tallen and David Paul Kane Educational & Research Foundation. T.P. was supported by ISF Grant 1208/04 and by a grant in Complexity Science from the Yeshaia Horvitz Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Hector Musto]
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Stern, A., Privman, E., Rasis, M. et al. Evolution of the Metazoan Protein Phosphatase 2C Superfamily. J Mol Evol 64, 61–70 (2007). https://doi.org/10.1007/s00239-006-0033-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0033-y