
Abstract
In mammals, males undergo a greater number of germline cell divisions compared with females. Thus, the male germline accumulates more DNA replication errors, which result in male mutation bias—a higher mutation rate for males than for females. The phenomenon of male mutation bias has been investigated mostly for rodents and primates, however, it has not been studied in detail for other mammalian orders. Here we sequenced and analyzed five introns of three genes (DBX/DBY, UTX/UTY, and ZFX/ZFY) homologous between X and Y chromosomes in several species of perissodactyls (horses and rhinos) and of primates. Male mutation bias was evident: substitution rate was higher for a Y chromosome intron than for its X chromosome homologue for all five intron pairs studied. Substitution rates varied regionally among introns sequenced on the same chromosome and this variation influenced male mutation bias inferred from each intron pair. Interestingly, we observed a positive correlation in substitution rates between homologous X and homologous Y introns as well as between orthologous primate and perissodactyl introns. The male-to-female mutation rate ratio estimated from concatenated sequences of five perissodactyl introns was 3.88 (95% CI = 2.90–6.07). Using the data generated here and estimates available in the literature, we compared male mutation bias among several mammalian orders. We conclude that male mutation bias is significantly higher for organisms with long generation times (primates, perissodactyls, and felids) than for organisms with short generation times (e.g., rodents) since the former undergo a greater number of male germline cell divisions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The number of germline cell divisions is greater for mammalian males than females (Vogel and Motulsky 1997). Thus, if mutations result from errors in DNA replication, mutation rate is expected to be higher in males than in females. Male mutation bias (a higher mutation rate in males than in females) for nucleotide substitutions has indeed been observed in several mammalian orders (Li et al. 2002) and led to the notion that mammalian evolution is male-driven. Male mutation bias was also shown for birds (Ellegren and Fridolfsson 1997) and fish (Ellegren and Fridolfsson 2003), as well as some plants (Filatov and Charlesworth 2002; Whittle and Johnston 2002).
Interestingly, the magnitude of male mutation bias (measured as α, or the male-to-female mutation rate ratio [Miyata et al. 1987]) differs among mammalian orders. Indeed, α ranges from ∼2 in rodents (Makova et al. 2004) to ∼5–6 in primates (Shimmin et al. 1993; Makova and Li 2002), with values that are intermediate in cats (Felidae, Carnivora [Pecon Slattery and O’Brien 1998]) and sheep and goats (Caprini, Cetartiodactyla [Lawson and Hewitt 2002]). It has been proposed that this variation can be explained by differences in generation time among different mammalian orders (Li et al. 2002), where generation time is defined as the average interval of time between the birth of an individual and the birth of its offspring. This can be explained by the following. While the generation time for rodents is shorter than that for primates, the number of germline cell divisions is approximately the same for primate and rodent females (Chang et al. 1994). Due to the longer primate generation time, however, primate males undergo a greater number of germline cell divisions compared with rodent males. Thus, the magnitude of male mutation bias is expected to correlate with generation time. However, additional data are required to test this hypothesis. This is not to be confused with the generation time effect hypothesis, according to which rodents have a higher substitution rate per time unit than primates because the former undergo a greater number of germline cell divisions per time unit compared with the latter (Laird et al. 1969).
Male mutation bias is usually assessed as the difference in substitution rates between the two sex chromosomes or between a sex chromosome and an autosome (Miyata et al. 1987). The ultimate way to analyze male mutation bias is from a genome-wide substitution rate comparison (Makova et al. 2004; Taylor et al. 2006). For those organisms lacking completely sequenced genomes, male mutation bias has been estimated from one (or a few) pair(s) of homologous loci located on different types of chromosomes (Li et al. 2002). These estimated values, however, can be influenced by regional substitution rate variation, which in turn depends on factors such as GC content, local recombination rate, and distance from origins of replication and telomeres (Hardison et al. 2003; Hellmann et al. 2005). As a result, the estimates of male mutation bias might be skewed. However, the extent to which male mutation bias can be influenced by regional variation in substitution rates is currently unknown.
In the current study we sequence five introns from three genes homologous between chromosome X and chromosome Y (genes homologous between sex chromosomes are called gametologues) in several species from two mammalian orders: Perissodactyla and Primates. We estimate the magnitude of male mutation bias and compare it among the introns, between these two orders, and with that of other mammalian orders (if available). Male mutation bias has not been previously investigated in the order Perissodactyla, whose members have a generation time intermediate to that of rodents and primates (although the ranges of generation times overlap between perissodactyls and primates). Furthermore, the influence of regional variation in substitution rates on male mutation bias has not been examined in any mammalian order. Specifically, this study addresses the following questions: (1) Does male mutation bias exist in perissodactyls? (2) Is male mutation bias in mammals influenced by generation time? (3) Does male mutation bias depend on regional variation in substitution rates?
We investigate the nucleotide substitution rate at introns of three gametologous genes: ZFX/ZFY, UTX/UTY, and DBX/DBY (also sometimes identified by DDX3X/DDX3Y). On human chromosome X, these genes are located relatively close to each other (Table 1) within the third evolutionary stratum of X-Y homology (Skaletsky et al. 2003). Recombination between X and Y ceased in that stratum ∼100 million years ago (MYA), before the radiation of eutherian mammals (Lahn and Page 1999). Thus, the X and Y copies of the selected introns do not recombine in either primates or perissodactyls. Human DBY and UTY cluster together close to the centromere on the long arm of Y (Yq), while ZFY is close to the telomere of the short arm of Y (Yp; Table 1). Although genomic sequences of perissodactyl sex chromosomes are not available, detailed physical mapping indicated conservation of gene order between horse and human X chromosomes (Raudsepp et al. 2002). In contrast, there is little conservation in gene order between horse and human Y chromosomes, however, UTY and DBY also cluster with each other in both species (Raudsepp et al. 2004).
Materials and Methods
Species analyzed
We analyzed five perissodactyls—Equus przewalskii (Przewalski’s horse), E. hemionus onager (onager), Diceros bicornis (black rhinoceros), Ceratotherium simum simum (white rhinoceros), and Tapirus indicus (Malayan tapir)—and five primates—Homo sapiens (human), Pongo pygmaeus (orangutan), Hylobates lar (white-handed gibbon), Colobus angolensis (Angolan colobus), and Saimiri boliviensis (Bolivian squirrel monkey). DNA samples for male and female specimens were available for all species except H. lar, C. angolensis, and S. boliviensis, for which we used male specimens only.
Amplification of Introns
One or two introns were amplifed from ZFX/ZFY, DBX/DBY, and UTX/UTY (Table 2). Amplification primers were designed with Oligo Lite V.6.66 (Molecular Biology Insights, Inc., Cascade, CO) as either universal, i.e., to amplify chromosomes Y and X introns together from males and chromosome X introns from females, or chromosome-specific, i.e., to amplify the X or Y copy of an intron separately. All primers (Supplemental Table 1) were designed based on the adjacent exon sequences of aligned H. sapiens X and Y genes. PCR products were column purified (Qiagen, Inc., Valencia, CA).
All primate amplifications were performed with chromosome-specific primers in 25-μl reactions that contained 50 ng genomic DNA, 1 × buffer, 0.2 mM dNTPs (PCR grade, Roche Applied Science, Indianapolis, IN), 0.7 U Expand High Fidelity PCR Enzyme mix (Roche Applied Science), 1.5 mM MgCl2, and 0.28 μM forward and reverse DNA primers (Integrated DNA Technologies, Inc., Skokie, IL). Cycling conditions are described in Supplemental Table 1.
X and Y amplification products for each of the perissodactyl male ZFX/ZFY and DBX/DBY introns were subcloned into the pCR2.1-TOPO vector with the TOPO TA Cloning kit (Invitrogen Life Technologies, Carlsbad CA). Plasmid inserts were amplified with pUC555 and pUC837 primers (Makova and Patton 1998) and Y-specific sequences were identified by comparison to X-specific sequences amplified from females. Perissodactyl UTY intron sequences were amplified with UTY11 forward and reverse primers (Hellborg and Ellegren 2003). All perissodactyl amplifications were performed in 2- μl reactions that contained 50 ng genomic DNA, 1 × buffer, 0.2 mM dNTPs (Gene Choice, Frederick, MD), 1 U Taq DNA polymerase (Invitrogen Life Technologies), 1.5 or 2.0 mM MgCl2, and 0.4 μM forward and reverse DNA primers (Integrated DNA Technologies, Inc). Two amplifications were performed for each individual per intron and the resulting products were sequenced. At least 10 clones were screened by sequencing for each cloned amplification.
Sequencing
All sequencing reactions were performed in 10-μl volumes and contained approximately 35 fmol purified PCR product, 1 μM sequencing primer, and 2 μl DTCS Quick Start Master Mix (Beckman Coulter, Inc., Fullerton, CA). Thermal cycling conditions were 96°C for 20 s, 50°C for 20 s, and 60°C for 4 min for 40 cycles. Sequencing products were purified by ethanol precipitation, then separated and analyzed on a CEQ8000 (Beckman Coulter, Inc.). All sequences generated here were deposited in GenBank (accession numbers DQ519372–DQ519376, DQ520642–DQ520730) Sequences of the last ZFX/ZFY intron for H. sapiens, P. pygmaeus, and S. boliviensis were downloaded from GenBank (accession numbers X58925, X58926, X58932, X72698, X58935, X58936).
Sequence Assembly and Alignments
Species- and chromosome-specific contigs for each intron were assembled in the SeqManII module of the Lasergene sequence analysis software (DNASTAR, Inc., Madison, WI). Heterozygous sites in female X sequences were resolved by randomly selecting one of the two represented bases. Intron-specific sequence alignments were made for the X or Y chromosome with ClustalW (Thompson et al. 1994) in the MegAlign module of the Lasergene software. Manual adjustments were made to alignments as needed.
Phylogenetic Analysis
Phylogenetic trees were constructed in MEGA3.1 (Kumar et al. 2004) for each X- and Y-specific intron and for each concatenation of all X- or Y-specific introns using the neighbor-joining method (Saitou and Nei 1987) with 1000 bootstrap replicas. Nucleotide substitution rates were estimated with the TN93 model (Tamura and Nei 1993). Branch-specific lengths were calculated in the baseml module of PAML (Yang 1997) using the TN93 model. Confidence intervals for substitution rates were calculated by nonparametric bootstrap (1000 replicates). The male-to-female mutation rate ratio (α) was calculated by substituting the ratio of the Y-specific (Y) and X-specific (X) branch lengths into the equation Y/X = 3α/(2 + α) (Miyata et al. 1987). The confidence intervals for α were estimated with nonparametric bootstrap (1000 replicates) as previously described (Bartosch-Harlid et al. 2003).
Permutation Tests
Permutation tests were performed to determine the statistical significance of substitution rate correlations observed between perissodactyl vs. primate introns on the same chromosome or between X vs. Y introns. In each such test we left the first variable intact and permuted the intron labels of the second variable. Namely, when perissodactyl and primate introns were compared, we did not change the labels of perissodactyl introns, but permuted intron labels of primate introns. Similarly, when X and Y chromosomes were compared, we did not change the labels of the X chromosome introns, but we permuted the labels of the Y chromosome introns. This was done exhaustively (5! = 120 times since we had five data points in each test). Fisher’s correlation coefficient (R) was calculated at each permutation. The empirical p value was calculated by comparison of the observed R with the distribution of R values from the permuted data sets.
Results
Sequences Obtained and Phylogenetic Analysis
We sequenced a total of ∼3–4 kb on chromosomes X and Y (Table 2) for each of five species of Perissodactyla and five species of primates. Five species representing all three known Perissodactyla families were analyzed: Przewalski’s horse and onager (Equidae), black rhinoceros and white rhinoceros (Rhinocerotidae), and tapir (Tapiridae). For comparison, we also analyzed five species from four primate families—human and orangutan (Hominidae), gibbon (Hylobatidae), colobus (Cercopithecidae), and squirrel monkey (Cebidae).
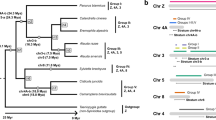
We built separate X- and Y-specific phylogenetic trees for perissodactyls or primates based on individual intron sequences (not shown) or on a concatenation of all intron sequences (Fig. 1). The topology of most trees corresponded to the phylogenetic relationship of the studied species obtained previously (Norman and Ashley 2000; Goodman et al. 2005), except that in several primate trees orangutan was grouped together with gibbon and not with human. Bootstrap support values were low for either human-orangutan or orangutan-gibbon clades. Human-orangutan grouping was assumed in the consequent analysis. The results did not change substantially depending on grouping within the human-orangutan-gibbon clade. Additionally, we built phylogenetic trees from X and Y sequences of each order aligned together (data not shown). For both perissodactyls and primates the topology within X- and Y-specific clades supported the accepted phylogeny of the studied species. This also indicated that gene conversion between X and Y gene copies was not evident in our data set. The analysis described below is based on separate X- and Y-specific phylogenetic trees.

Male mutation bias for perissodactyls (A) and primates (B) from concatenated sequences of five gametologous intron pairs. The numbers above each branch are the numbers of nucleotide substitutions per 100 sites estimated from the Y-linked and the X-linked intron sequences, respectively. The number below each branch is the male-to-female mutation rate ratio (α). The branch lengths are not drawn precisely to scale.
Regional Variation in Substitution Rates
Substantial variation in substitution rates was observed even when introns sequenced for the same chromosome and within the same order were compared to each other (Table 2). On average, any two introns within each of these four groups (primate Y introns, primate X introns, perissodactyl Y introns, and perissodactyl X introns) differed in their substitution rates 1.5 times. However, occasionally the difference in substitution rates between such introns was as high as three times (e.g., between next to the last intron of ZFX [ZFX-ntl] and DBX-9 intron sequenced in perissodactyls).
Substitution rates were similar between genomic regions separated by a small physical distance on a chromosome. Introns of the same gene (when analyzed for the same order) had similar substitution rates (Table 2). Indeed, only two comparisons (of a total of eight) between introns of the same gene (either DBX/DBY or ZFX/ZFY) resulted in significantly different substitution rates (as measured by non-overlapping 95% confidence intervals). The two introns sequenced for the same gene were separated by less than 1.2 kb (Table 1). Substitution rates between UTX (or UTY) and either of the two DBX (or DBY) introns were not significantly different (overlapping 95% confidence intervals). The distance between UTY and DBY is only ∼0.3 Mb and the distance between UTX and DBX is ∼3.5 Mb. In contrast, approximately half (11 of 24) of the comparisons between either of the two ZFX (or ZFY) introns and individual introns of the other two genes resulted in significantly different substitution rates. ZFX is located ∼17 and ∼21 Mb away from DBX and UTX, respectively, and ZFY is situated on a chromosomal arm opposite that of DBY and UTY (Table 1).
We next investigated whether there was a correlation in substitution rates between perissodactyl and primate orthologous introns. Although we analyzed only five introns in each comparison, a positive correlation in substitution rates was indeed observed between primate and perissodactyl orthologous introns located either on chromosome X (R = 0.59, p = 0.10, significance assessed by a permutation test) or on chromosome Y (R = 0.66, p = 0.10). Interestingly, we also observed a positive correlation in intron substitution rates between gametologous introns within either perissodactyls (R = 0.75, p = 0.07) or primates (R = 0.95, p = 0.03).
GC content has been shown to determine at least part of the variation in substitution rates (Hardison et al. 2003). Contrary to this, GC content and intron substitution rate were not correlated in our data. For instance, the two DBX introns sequenced in primates had almost-identical substitution rates (∼0.16) but differed drastically in their GC content (31% for DBX6 and 19% for DBX9). We did not investigate the relationship between substitution rates and recombination rates because the latter rates are not available for perissodactyls and, moreover, are known to vary even between closely related species in primates (Ptak et al. 2004).
Male Mutation Bias
The substitution rate was higher for a Y chromosome intron than for its X chromosome counterpart in all five pairs of gametologous introns (either within primates or within perissodactyls), consistent with the male mutation bias hypothesis (Table 2). The α values differed substantially among individual perissodactyl introns (from 1.92 to 31.8) as well as among individual primate introns (from 2.59 to 10.8). Note that such differences were rarely significant due to wide confidence intervals of α values for each gametologous intron pair. Indeed, the α estimates of all primate introns had overlapping 95% confidence intervals, and in perissodactyls only α values compared between DBX-9/DBY-7 and either ZFX-ntl/ZFY-ntl or DBX-6/DBY-4 introns were significantly different.
Due to the aforementioned variation among introns, the most reliable α values were obtained from sequences concatenated either for X or for Y for each order. This led to α = 3.88 (95% CI = 2.90–6.07) and α = 3.79 (95% CI = 2.71–5.99) for perissodactyls and primates, respectively. Branch-specific α values displayed considerable variation (Fig. 1). In particular, short external branches leading to the two horse species and to the two rhinoceros species (a total of four branches) gave very different estimates of α. Within each of these two pairs of branches, one α value was close to 1 and the other one was infinity. As the two studied species are closely related in each of these two pairs (Oakenfull and Clegg 1998; Norman and Ashley 2000), the magnitude of male mutation bias estimated for these external branches might be affected by ancient genetic diversity. Indeed, for closely related species, diversity at the time of speciation constitutes a significant portion of the observed divergence (Li 1977). Differences in diversity between the two sex chromosomes might lead to biased α values (Makova and Li 2002). Exclusion of the four short external branches led to α = 2.97 for perissodactyls. Closely related species were absent from our primate data set.
Discussion
Our analysis of male mutation bias for five introns of three genes gametologous between X and Y chromosomes in two mammalian orders led to the following findings and conclusions. First, in agreement with other studies (e.g., Lercher et al. 2001; Hardison et al. 2003; Gaffney and Keightley 2005), we observed considerable intrachromosomal (regional) variation in substitution rates among different introns sequenced on the same chromosome. Substitution rates were similar for introns within the same gene and between genes separated by a small distance (DBX/DBY and UTX/UTY) and were usually different between genes separated by a large distance (ZFX/ZFY vs. DBX/DBY and UTX/UTY), echoing an earlier finding that regional variation is determined by local features of the genome (Hardison et al. 2003). Our results also agree well with the study by Gaffney and colleagues (Gaffney and Keightley 2005), who found that similarity in murid substitution rates was at its maximum under 100 kb, decreased substantially at ∼5 Mb, and was already undetectable at ∼15 Mb.
Interestingly, a positive correlation in substitution rates was observed between perissodactyl and primate orthologous introns as well as between X and Y introns within the same order. This similarity in substitution rates among orthologous and gametologous introns may be attributed to similarities in sequence content and other genomic/chromosomal factors probably inherited from the common ancestor – either the perissodactyl-primate common ancestor that existed ∼95 MYA (Springer et al. 2003) or the stratum 3 pseudoautosomal ancestor that existed more than ∼100 MYA (Lahn and Page 1999). A positive correlation was also observed between rodent-specific and rodent-primate local substitution rates in another study (Gibbs et al. 2004). Rodents and primates are thought to have diverged ∼90 MYA (Springer et al. 2003).
Second, regional variation in substitution rates on the two sex chromosomes in turn led to variation in the estimated magnitude of male mutation bias obtained from different introns. The resulting α values sometimes differed as much as 16 times (e.g., when compared between ZFX-ntl/ZFY-ntl and DBX-9/DBY-7 in perissodactyls)! Thus, the estimates of male mutation bias obtained from sequences with different genomic locations (and different regional factors determining substitution rates) might not be directly comparable to each other (see also Taylor et al. 2006).
Third, we obtained a new estimate of male mutation bias in primates and the first such estimate in perissodactyls, and provided support of the correlation between male mutation bias and generation time in mammals. The primate α = 3.79 (95% CI = 2.71–5.99) obtained here was somewhat lower but not significantly different from other primate α estimates based on X–Y comparison (excluding the ones derived from closely related species, e.g. human-chimpanzee comparison) which ranged from 4.20 to 6.26 and usually had wide confidence intervals (Shimmin et al. 1993; Chang and Li 1995; Huang et al. 1997). Data generated here nearly doubled the available sequences to estimate male mutation bias in primates, again excluding comparisons between closely related species because such comparisons might be influenced by ancient genetic diversity (Makova and Li 2002).
The magnitude of male mutation bias for Perissodactyla estimated here (α = 3.88) was similar to that observed for Felidae (Carnivora) and Caprini (Cetartiodactyla; Fig. 2). The studied species of these three taxonomic groups have similar generation times (Supplemental Table 2). The estimate of α in perissodactyls was also similar to that obtained in the present study for primates, even though on average primates have longer generation times compared with perissodactyls. This can be explained by overlapping 95% confidence intervals of α as well as by overlapping generation time ranges among Perissodactyla and Primates (Fig. 2). It would be informative to increase the number of analyzed bases for both perissodactyls and primates to narrow the confidence intervals for α estimates.

Relationship between male mutation bias and male age at first reproduction (used as an approximation of generation time). The 95% confidence intervals for α are represented by dashed lines and the ranges of male age at first reproduction (Supplemental Table 2) are represented by solid lines. The sources of data on male mutation bias are as follows: rodents—Makova et al. (2004); felids—Pecon Slattery and O’Brien (1998); Caprini—Lawson and Hewitt (2002); Perissodactyla—this study; primates—this study and pooled data from Shimmin et al. (1993), Chang and Li (1995), and Huang et al. (1997).
A distinct feature of our results is that rodents, which have a short generation time, had significantly weaker male mutation bias than Primates, Perissodactyla, Caprini, and Felidae, which have relatively long generation times. Thus, an analysis of five mammalian orders indicates stronger male mutation bias in organisms with longer generation time (Fig. 2), in agreement with the conclusions of the study investigating the same relationship in birds (Bartosch-Harlid et al. 2003). Here we assumed a star-like phylogeny among the analyzed mammalian orders to avoid statistical dependence due to shared phylogenetic history. While this is a simplifying assumption, it has been accepted that radiation of the orders of eutherian mammals occurred rapidly (Springer et al. 2003).
For Felidae (Pecon Slattery and O’Brien 1998) and Caprini (Lawson and Hewitt 2002), the currently available estimates of male mutation bias are still based on the analysis of just one locus—ZFX/ZFY. However, the analysis of ZFX/ZFY might not represent the ultimate way to compare male mutation bias among mammalian orders because the physical position of ZFY has not been conserved throughout eutherian evolution (Raudsepp et al. 2004). Thus the substitution rate at this locus might depend on different regional factors for each order. For mammalian orders other than the five discussed above we have no male mutation bias information.
Here we approximate generation time by male age at first reproduction (Supplemental Table 2; Nowak 1991). We are taking male and not female age at first reproduction in particular because the former is expected to determine the number of male germline cell divisions—the number that differs among mammalian orders (at least between human and mouse or rat [Chang et al. 1994]). The number of female germline cell divisions is likely to be constant among mammalian orders (again judging from the comparison of human and rodent female reproductive physiology [Chang et al. 1994]). Data on the number of germline cell divisions in nonmodel mammalian species are critically needed for the studies of male mutation bias.
Besides data on the number of germline cell divisions, genomic sequences for at least two species (that are not closely related) per mammalian order are required to estimate male mutation bias accurately and further test its correlation with generation time. One such species pair is already available (mouse and rat) and male mutation bias has been investigated in it (Makova et al. 2004). Soon a similar opportunity will arise to obtain a reliable estimate of male mutation bias in primates from the human-macaque comparison. Note that in most genome-wide studies male mutation bias can be estimated more reliably from the X-autosomal comparison due to either unavailability (females being sequenced) or incomplete sequence of chromosome Y because of its highly repetitive and palindromic nature (Skaletsky et al. 2003). Targeted sequencing of individual Y chromosomes such as the one undertaken for human and its comparison with Y chromosomes from species of the same order are expected to provide the “Y side” of the male mutation bias story.
References
Bartosch-Harlid A, Berlin S, Smith NG, Moller AP, Ellegren H (2003) Life history and the male mutation bias. Evol Int J Org Evol 57:2398–2406
Chang BH, Li WH (1995) Estimating the intensity of male-driven evolution in rodents by using X-linked and Y-linked Ube 1 genes and pseudogenes. J Mol Evol 40:70–77
Chang BH, Shimmin LC, Shyue SK, Hewett-Emmett D, Li WH (1994) Weak male-driven molecular evolution in rodents. Proc Natl Acad Sci USA 91:827–831
Ellegren H, Fridolfsson AK (1997) Male-driven evolution of DNA sequences in birds. Nat Genet 17:182–184
Ellegren H, Fridolfsson AK (2003) Sex-specific mutation rates in salmonid fish. J Mol Evol 56:458–463
Filatov DA, Charlesworth D (2002) Substitution rates in the X- and Y-linked genes of the plants, Silene latifolia and S. dioica. Mol Biol Evol 19:898–907
Gaffney DJ, Keightley PD (2005) The scale of mutational variation in the murid genome. Genome Res 15:1086–1094
Gibbs RA, Weinstock GM, Metzker ML, et al. (2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428:493–521
Goodman M, Grossman LI, Wildman DE (2005) Moving primate genomics beyond the chimpanzee genome. Trends Genet 21:511–517
Hardison RC, Roskin KM, Yang S, Diekhans M, Kent WJ, Weber R, Elnitski L, Li J, O’Connor M, Kolbe D, Schwartz S, Furey TS, Whelan S, Goldman N, Smit A, Miller W, Chiaromonte F, Haussler D (2003) Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res 13:13–26
Hellborg L, Ellegren H (2003) Y chromosome conserved anchored tagged sequences (YCATS) for the analysis of mammalian male-specific DNA. Mol Ecol 12:283–291
Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE (2005) Why do human diversity levels vary at a megabase scale? Genome Res 15:1222–1231
Huang W, Chang BH, Gu X, Hewett-Emmett D, Li W (1997) Sex differences in mutation rate in higher primates estimated from AMG intron sequences. J Mol Evol 44:463–465
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163
Lahn BT, Page DC (1999) Four evolutionary strata on the human X chromosome. Science 286:964–967
Laird CD, McConaughy BL, McCarthy BJ (1969) Rate of fixation of nucleotide substitutions in evolution. Nature 224:149–154
Lawson LJ, Hewitt GM (2002) Comparison of substitution rates in ZFX and ZFY introns of sheep and goat related species supports the hypothesis of male-biased mutation rates. J Mol Evol 54:54–61
Lercher MJ, Williams EJ, Hurst LD (2001) Local similarity in evolutionary rates extends over whole chromosomes in human-rodent and mouse-rat comparisons: implications for understanding the mechanistic basis of the male mutation bias. Mol Biol Evol 18:2032–2039
Li WH (1977) Distribution of nucleotide differences between two randomly chosen cistrons in a finite population. Genetics 85:331–337
Li WH, Yi S, Makova K (2002) Male-driven evolution. Curr Opin Genet Dev 12:650–656
Makova KD, Li WH (2002) Strong male-driven evolution of DNA sequences in humans and apes. Nature 416:624–626
Makova KD, Patton JC (1998) Increased yield of tri- and tetranucleotide heterospecific microsatellites from unenriched small-insert libraries. Biotechniques 24:38, 40, 42–43
Makova KD, Yang S, Chiaromonte F (2004) Insertions and deletions are male biased too: a whole-genome analysis in rodents. Genome Res 14:567–573
Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T (1987) Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol 52:863–867
Norman JE, Ashley MV (2000) Phylogenetics of Perissodactyla and tests of the molecular clock. J Mol Evol 50:11–21
Nowak RM (1991) Walker’s mammals of the world. John Hopkins University Press, Baltimore
Oakenfull EA, Clegg JB (1998) Phylogenetic relationships within the genus Equus and the evolution of alpha and theta globin genes. J Mol Evol 47:772–783
Pecon Slattery J, O’Brien SJ (1998) Patterns of Y and X chromosome DNA sequence divergence during the Felidae radiation. Genetics 148:1245–1255
Ptak SE, Roeder AD, Stephens M, Gilad Y, Paabo S, Przeworski M (2004) Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biol 2:849–855
Raudsepp T, Kata SR, Piumi F, Swinburne J, Womack JE, Skow LC, Chowdhary BP (2002) Conservation of gene order between horse and human X chromosomes as evidenced through radiation hybrid mapping. Genomics 79:451–457
Raudsepp T, Santani A, Wallner B, Kata SR, Ren C, Zhang HB, Womack JE, Skow LC, Chowdhary BP (2004) A detailed physical map of the horse Y chromosome. Proc Natl Acad Sci USA 101:9321–9326
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Shimmin LC, Chang BH, Li WH (1993) Male-driven evolution of DNA sequences. Nature 362:745–747
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, Chinwalla A, Delehaunty A, Delehaunty K, Du H, Fewell G, Fulton L, Fulton R, Graves T, Hou SF, Latrielle P, Leonard S, Mardis E, Maupin R, McPherson J, Miner T, Nash W, Nguyen C, Ozersky P, Pepin K, Rock S, Rohlfing T, Scott K, Schultz B, Strong C, Tin-Wollam A, Yang SP, Waterston RH, Wilson RK, Rozen S, Page DC (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837
Springer MS, Murphy WJ, Eizirik E, O’Brien SJ (2003) Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc Natl Acad Sci USA 100:1056–1061
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Taylor J, Tyekucheva S, Zody M, Chiaromonte F, Makova KD (2006) Strong and weak male mutation bias at different sites in the primate genomes: insights from the human-chimpanzee comparison. Mol Biol Evol 23:565–573
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vogel F, Motulsky AG (1997) Human genetics: problems and approaches. Springer, Berlin
Whittle CA, Johnston MO (2002) Male-driven evolution of mitochondrial and chloroplastidial DNA sequences in plants. Mol Biol Evol 19:938–949
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Acknowledgments
We thank Matt Ghent for help in the analysis, Chungoo Park for implementing the permutation test, and Erika Kvikstad for critical reading of the manuscript. The perissodactyl DNA samples were kindly provided by the San Diego Zoological Society. This study was supported by NIH Grant R01-GM072264 and by start-up funds from the Eberly College of Sciences at Penn State University to K.D.M.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Deborah Charlesworth]
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Goetting-Minesky, M.P., Makova, K.D. Mammalian Male Mutation Bias: Impacts of Generation Time and Regional Variation in Substitution Rates. J Mol Evol 63, 537–544 (2006). https://doi.org/10.1007/s00239-005-0308-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0308-8