
Abstract
The genome organization of the Hardness locus in the tribe Triticeae constitutes an excellent model for studying the mechanisms of evolution that played a role in the preservation and potential functional innovations of duplicate genes. Here we applied the nonsynonymous-synonymous rate ratio (d N /d S or ω) to measure the selective pressures at the paralogous puroindoline-a (Pina), puroindoline-b (Pinb), and grain softness protein-1 (Gsp-1) genes located at this locus. Puroindolines represent the molecular-genetic basis of grain texture. In addition, the puroindoline gene products have antimicrobial properties with potential role in plant defense. We document the complete coding sequences from the Triticum/Aegilops taxa, rye and barley including the A, D, C, H, M, N, R, S, and U genomes of the Triticeae. Maximum likelihood analyses performed on Bayesian phylogenetic trees showed distinct evolutionary patterns among Pina, Pinb, and Gsp-1. Positive diversifying selection appeared to drive the evolution of at least one of the three genes examined, suggesting that adaptive forces have operated at this locus. Results evidenced positive selection (ω > 4) at Pina and detected amino acid residues along the mature PIN-a protein with a high probability (>95%) of having evolved under adaptation. We hypothesized that positive selection at the Pina region is congruent with its role as a plant defense gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene duplication is a key mechanism in the evolution of gene function because once a gene is duplicated redundant copies can accumulate mutations and diverge to create evolutionary innovations (Ohno 1970). It is not clear, however, whether functional divergence of duplicates arise by fixation of advantageous mutation (positive Darwinian selection) or by random fixation of selectively neutral mutations (genetic drift) (Clark 1994; Lynch et al. 2001; Moore and Purugganan 2003).
The homologous puroindoline-a (Pina), puroindoline-b (Pinb), and grain softness protein-1 (Gsp-1) genes located at the Hardness locus in the Pooideae subfamily have likely originated from two independent duplication events from an ancestral Pooideae Gsp-1 gene (Tranquilli et al. 1999; Chantret et al. 2004). Evidence for additional gene duplication has been reported in the Triticum/Aegilops and Hordeum genera involving puroindoline-b (“relic” and “pseudogene”) and the homologous hordoindoline-b gene (Hinb-1 and Hinb-2) (Beecher et al. 2001; Darlington et al. 2001; Caldwell et al. 2004; Chantret et al. 2005).
Although gene duplication has been well documented, little is known about the evolutionary dynamics that shaped sequence diversity in Pina, Pinb, and Gsp-1 after duplication events (Massa et al. 2004). Here, we identify, annotate, and analyze the complete protein coding region of the three genes from a wide range of diploid Triticum/Aegilops genomes, rye and barley, and inferred phylogenetic relationships as a basis to determine which genes underlie molecular adaptation and what selective pressures have driven their evolution.
Positive Darwinian selection is of particular interest because adaptive changes in genes are eventually responsible for evolutionary novelties. One approach for detecting selection in a protein coding region is through the ratio of nonsynonymous (d N , replacement) to synonymous (d S , silent) mutation rates d N /d S , also called ω (Miyata et al. 1979; Li et al. 1985). Thus, d N /d S < 1, d N /d S = 1, and d N /d S > 1 represent negative (purifying) selection, neutral evolution, and positive (adaptive) selection, respectively. However, since most proteins have highly conserved regions or specific amino acid residues, where replacement mutations are not tolerated (with d N close to 0), the comparison of any pair of genes may fail to detect positive selection when averaging the d S and d N rates over all sites. Alternative methods that account for variable selective pressure across sites have been applied to detect adaptive evolution in a number of protein-coding genes (Nielsen and Yang 1998; Yang et al. 2000).
Grain softness protein-1 (GSP-1) and the two isoforms of puroindoline proteins (PIN-a and PIN-b) are members of the 2S family of basic and cysteine-rich proteins (Shewry and Morell 2001). First isolated from wheat endosperm, they have been reported in several other taxa of the tribe Triticeae including rye and barley (Gautier et al. 2000; Lillemo et al. 2002; Massa et al. 2004). In T. aestivum, an allohexaploid (2n = 6x = 42 AABBDD genomes) which combined the AB genome of tetraploid Triticum turgidum L. ssp. dicoccum Shrank ex Schübler with the diploid D genome of Ae. tauschii, the puroindoline loci from the A- and B-genomes were lost during the initial tetraploidation event, but not Gsp-1 (Gautier et al. 2000; Ozkan et al. 2001). Consequently, T. aestivum contains three homoeologous Gsp-1 loci (Gsp-A1, Gsp-B1, and Gsp-D1) but only a single locus each for the puroindolines (Pina-D1 and Pinb-D1).
PINs are unique among plant proteins because of their tryptophan-rich hydrophobic domain. Interestingly, two different biological functions have been proposed for PINs. First, they constitute the molecular basis of grain endosperm texture, which is a primary determinant of end-use quality of wheat (Giroux and Morris 1997, 1998; Morris 2002). With both puroindolines in their functional form, the endosperm is soft and friable. However, when either one of the puroindolines is absent or mutated, the endosperm is hard textured (Giroux and Morris 1997, 1998; Morris 2002). Second, they have been proposed as antimicrobial proteins that play a role in seed protection (Dubreil et al. 1998). Functional assays in vivo (Krishnamurthy et al. 2001) and in vitro (Dubreil et al. 1998; Le Guerneve et al. 1998; Jing et al. 2003) have demonstrated the antimicrobial activity of both PINs against plant pathogens. It has been suggested that PINs act synergistically with thionins and exert their antifungal-antimicrobial properties through the interaction with lipid cell membranes (Dubreil et al. 1998; Le Guerneve et al. 1998; Jing et al. 2003).
Here we show that Pina, Pinb, and Gsp-1 genes have distinct rates of sequence evolution, favoring the hypothesis that they have been subjected to different selective constraints since the events of gene duplication. We found significant statistical support for the presence of amino acid positions along the Pina sequence with a high probability of having been fixed by natural selection, suggesting that adaptive forces have governed the evolutionary dynamics of this region. We hypothesized that patterns of Pina evolution are consistent with its role as a plant defense gene. In addition, our study detected target DNA sequences and amino acid residues that are worthy of further experimental functional analyses.
Materials and Methods
Pina, Pinb, and Gsp-1 genes were examined in the tribe Triticeae including all diploid species of the genus Aegilops (van Slageren 1994), Triticum monococcum L., Triticum urartu Thüm. ex Gandilyan, Secale cereale L. (rye) cv. Galma, and Hordeum vulgare L. subsp. vulgare (barley) cv. Morex (Table 1). DNA sequences of the Pina, Pinb, and Gsp-1 genes from Ae. tauschii and the wild-type alleles from T. aestivum have been reported previously (Massa et al. 2004). For comparison, we also included the homoeologous Gsp-1 genes (Gsp-A1, Gsp-B1, and Gsp-D1) of T. aestivum cv. Chinese Spring. The Gsp-1 genome-specific sequences of T. aestivum were retrieved from GenBank dbEST using the Gsp-D1 of T. aestivum cv. Yecora Rojo, accession AY255771 (Massa et al. 2004), as a query in BLAST searches. The identified ESTs were then assigned to the A, B, and D genomes using the 5AS-3, 5BS-6, and 5DS-2 deletion lines of Chinese Spring (Endo and Gill 1996), respectively. To further increase statistical support for phylogenetic trees and codon-based maximum likelihood estimates, we incorporated publicly available DNA sequences from diploid and tetraploid Aegilops species (AY608592, AY608594, AY608599) and barley (AY643843, AY644302) (Table 1).
DNA Analysis
Isolation and amplification of genomic DNA were performed as described by Massa et al. (2004), except that the present study included additional primers (Table 2). Gene- and genome-specific primers were primarily designed to flank closely the coding region. Furthermore, based on locus-specific polymorphisms, we designed SNP primers to amplify the homoeologous Gsp-1 sequences in Chinese Spring (Table 2). The annealing temperature of PCR was optimized for each primer combination, and the PCR-amplification products were either sequenced directly or cloned into a pCR 2.1 cloning vector (TA Cloning kit, Invitrogen, Carlsbad, CA). At least three independent clones per gene were sequenced when needed to resolve ambiguities.
Molecular Evolution and Phylogenetic Analyses
Multiple sequence alignments were performed using ClustalW (Thompson et al. 1994). Gene trees were generated by maximum likelihood (ML) algorithms as implemented in PAUP* 4.0 beta 10 (Swofford 2002) and by Bayesian inferences as performed in MrBayes 3.1 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). The nucleotide substitution models used in both ML and Bayesian methods were estimated by the Akaike Information Criterion in Modeltest 3.6 (Posada and Crandall 1998). Three models were selected: K81uf+G (for Pina), TVMef+G (for Pinb), and K80+G (for Gsp-1). For ML, the tree topologies were evaluated by heuristic searches with random-addition-sequence replications and tree bisection-reconnection branch swapping. The support of clades was assessed with bootstrapping with 500 replicates. In Bayesian phylogenetic reconstructions, two simultaneous Markov chain Monte Carlo analyses were run for about 2.5 × 106 generations starting from different random trees. The closest available substitution models were applied accordingly, that is, K81uf+G (nst = 6, rates = gamma), TVMef+G (nst = 6, rates = gamma), and K80+G (nst = 2, rates = gamma). The 50% majority-rule consensus trees were obtained after excluding 25% of the samples as “burn-in” (MrBayes 3.1).
The codon-based maximum likelihood method was applied to estimate the nonsynonymous (d N )/synonymous (d S ) rate ratio (d N /d S = ω). Calculations were conducted using the CODEML program in PAML 3.14 software package (Yang 1997). Under the site-specific models, which allow the ω ratio to vary among sites (Nielsen and Yang 1998; Yang et al. 2000), we implemented likelihood ratio tests (LRTs) to compare two pairs of nested models. That is, we computed twice the difference between the log-likelihoods, 2Δℓ = 2 × (ℓ0 − ℓ1), to compare M1a (nearly neutral) against M2a (positive selection), and M7 (ß) against M8 (ß&ω), both using a chi-square distribution with two degrees of freedom (df). A Bayes empirical Bayes (BEB) approach was then used to calculate the posterior probability (P b ) that a site is under positive selection (Wong et al. 2004; Yang et al. 2005).
Since ML methods based on models of codon substitution do not account for the effects of recombination, we subjected our data sets to two different tests: the maximum chi-square test of Maynard Smith (1992) and the detection method of gene conversion implemented in GENECONV 1.81 (Sawyer 1989).
Results
The full-length of the Pina, Pinb, and Gsp-1 genes was obtained for all diploid Triticum/Aegilops species. We also amplified Gsp-1, secaloindoline-a, and secaloindoline-b from rye and hordoindoline-a (Hina) from barley. Since the same primers were used for all taxa, the amplification of genes from distant species like Triticum and Hordeum suggests that conserved nucleotide sequences should exit at the primer binding sites.
The data set of Pina comprised 31 sequences from 17 taxa including three accessions retrieved from GenBank: AY608599 (Ae. biuncialis), AY608592 (Ae. kotschyi), and AY608594 (Ae. caudata). The aligned region was 450 bp long, with a total number of 438 sites (excluding gaps, 146 codons). Of these, 342 were invariant (78%) and 96 (22%) were variable (polymorphic) sites.
For Pinb we included 33 sequences from 15 taxa. The two putative paralogous genes from barley (Hinb-1 and Hindb-2) were recovered from GenBank (AY643843). The sequence length was 450 bp, with a total number of 438 sites (excluding gaps, 146 codons). About 71% (313) of the total sites were invariant and 29% (125) were variable. The Gsp-1 data set consisted of 32 sequences from 15 taxa and included the homoeologous Gsp-1 genes from the A, B, and D genomes of T. aestivum cv. Chinese Spring. The sequences were 492 bp long, with a total number of 489 sites (163 codons) after removing gaps. Of these, 357 sites (73%) were invariant and 132 sites (27%) were polymorphic.
All sequences obtained here were deposited in GenBank under accession numbers DQ269819–DQ269918 (Table 1). Some sequences were identical, and only different sequences were used in later analyses.
Sequence Diversity in the Tryptophan-Rich Domain of Pina and Pinb
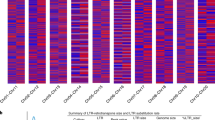
Variation in the tryptophan-rich domain is of particular importance to the structure and function of both PIN-a and PIN-b proteins. Nucleotide and predicted amino acid sequence comparisons showed a highly conserved Pinb domain (WPTKWWK) both within and between species (Fig. 1). However, we identified three nonsynonymous substitutions (amino acid-altering) mutations (R65Q, K68R, and K68T) in the tryptophan-rich region (WRWWKWWK) of PIN-a (Fig. 1). Note that in the present study the numbering of amino acid or codon sites is based on the metheonine (M) start codon as 1 (Fig. 1). The lysine-to-arginine change at position 68 (K68R) was common to all S-genome Aegilops species, whereas the other two mutations involved radical changes that might affect the molecular structure of the protein. The arginine-to-glutamine change at position 65 (R65Q) was identified in secaloindoline-a of S. cereale cv. Galma and is reported here for the first time; while the putative lysine-to-threonine mutation at position 68 (K68T) detected in H. vulgare L. subsp. vulgare cv. Morex is common to several barley cultivars (publicly available DNA sequences), suggesting that it may be a species-specific mutation.

Sequence alignment of the Pina region containing the codon sites identified as being under positive selection (indicated by rectangles). Residues 78–145 are not shown. The sequences are arranged by phylogenetic groups as in Fig. 2. Genome abbreviations are on the left. A dot shows identity with the first sequence. A dash indicates a gap, which was inserted in the sequence to preserve the alignment. Amino acid numbering is based on the metheonine (M) start codon as 1. Gaps are ignored.
Molecular Phylogenies
ML and Bayesian inference algorithms produced robust phylogenies for all three genes studied. The tree topologies created by both methods were similar in their overall structure but only Bayesian trees are presented (Figs. 2–5). In addition to the single-gene genealogies, we also constructed a global phylogeny, which regrouped the three genes (Fig. 2). The global tree based on 96 sequences clearly distinguished the paralogous Pina, Pinb, and Gsp-1 genes and inferred putative orthologous relationships within each gene cluster. The results also indicated the cluster of the paralogous Hinb-1 and Hinb-2 genes (94% amino acid similarity), suggesting that duplication must have occurred after the divergence of Hordeum from the Triticum/Aegilops lineages (Figs. 2 and 4).

Bayesian phylogeny based on 96 sequences from Pina, Pinb, and Gsp-1 genes. Genome clusters (solid triangles) are indicated with the same letter as listed in Table 1.The size is proportional to the number of sequences. Numbers above branches represent Bayesian posterior probabilities of finding a given clade (only those of the major gene clusters are indicated). Scale bar, 0.1 nucleotide substitution.

Bayesian phylogeny of the Pina gene based on 31 distinct sequences of 17 taxa of the tribe Triticeae. Haplotypes are labeled by species name and accession identifiers (in parentheses). Genome abbreviations are the same as in Fig. 1. Numbers above branches represent both posterior probabilities and bootstrap values from the ML analysis. Branch lengths were reestimated on the initial Bayes tree topology under the codon-substitution model and calculated by the expected number of nucleotide substitutions per codon. Scale bar, 0.1 substitution per codon.

Bayesian phylogeny of the Pinb gene based on 33 distinct sequences of 15 taxa of the tribe Triticeae. Tree description and labels are the same as in Fig. 2.

Bayesian phylogeny of the Gsp-1 gene based on 32 distinct sequences of 15 taxa of the tribe Triticeae. Tree description and labels are the same as in Fig. 2.
Single-gene genealogies were primarily resolved based on genome-specific mutations. Bootstrap and posterior probabilities supported the majority of genome-specific nucleotide changes across taxa (>75% and >0.75, respectively). Consequently, unique mutations could be traced through the Triticum/Aegilops evolution. For example, the homoeologous Gsp-1 loci of hexaploid wheat (i.e., Gsp-A1, Gsp-B1, and Gsp-D1) defined known relationships with the corresponding A, S, and D genomes of T. urartu (Au Au), Ae. speltoides (SS), and Ae. tauschii (DD), respectively (Fig. 5). Likewise, the U-genome Pina sequences of the tetraploids Ae. biuncialis (UUMM) and Ae. kotschyi (UUSS), grouped together with the U genome of Ae. umbellulata (UU) (Fig. 3). Similarly, the S-genome species, including those with the S b, S l, and S s subgenomes, were unambiguously supported as a monophyletic group in all three gene phylogenies (Figs. 2–5). The only exception was the Pinb alleles of Ae. tauschii (D genome), which did not coalesce within the species; actually, two alleles of Ae. tauschii (TA2450 and TA2536) were more closely related to other Triticum/Aegilops alleles than they were to TA1583 and TA1599 (Fig. 4). These results are consistent with our previous observations that polymorphism at this locus have persisted through speciation events (transspecies polymorphism) (Massa et al. 2004).
Although phylogenetic relationships inferred from single-gene loci were well resolved, comparisons among gene trees from different loci suggested phylogenetic disagreements. In principle, conflict among trees can be due to assortment of ancestral polymorphism or homoplasy. These important factors, if present, could confound the signal of selection. For example, strong positive selection could lead to homoplasy at sites under selection and this could lead to a gene that reflects the effects of this selection/homoplasy rather than the actual historical relationships among alleles. Since ML analyses require the use of the true phylogeny/genealogy, when the tree used is incorrect, inference of selection may be sensitive to the assumed tree topology (Yang et al. 2000). One way to look at this issue has been to use a consensus tree or a putative species tree. However, there is no means to justify that any given set of relationships among the Triticum/Aegilops lineages is the correct one (R. Mason-Gamer, personal communication). In this study, we examined the effects of tree topology by analyzing the same data with a few candidate trees (see next section).
Gene Evolution
To elucidate whether Pina, Pinb, and Gsp-1 have been subjected to positive selection (ω > 1) we applied the codon-based substitution models as implemented in the CODEML program of the PAML software package. LRTs and prediction of sites under positives selection (BEB inference) were performed on both ML and Bayesian gene trees, although only the results with the best likelihood scores are presented (Figs. 3–5). We fixed the branch lengths at those which were calculated under the one-ratio model (M0) and used them when running the M1a (nearly neutral), M2a (positive selection), M7 (ß), and M8 (ß&ω) models.
The best tree for Pina, according to the one-ratio model (M0), was the corresponding gene tree based on Bayesian analysis (Fig. 3). The M2a and M8 models predicted the occurrence of positively selected sites (ω > 1) and showed ML values higher than their corresponding null models, M1a and M7, respectively (Table 3). The LRT statistics were 2Δℓ = 11.04 for the comparison of M1a and M2 and 2Δℓ = 12.42 for the comparison of M7 and M8. Both rejected the null models with p < 0.01 when compared with a chi-square distribution with 2 df. Although many amino acid sites appear to be under strong purifying selection (>78%), results indicated that ∼6% of the codon sites evolved under adaptive evolution with an average ω ratio >4 (Table 3). Under the M8 model, the BEB inference identified two positively selected sites, 28V and 39V, with a posterior probability p b ≥ 95%, and 75Q with a p b = 94.4% (Table 3). Similarly, the BEB inference detected the same sites under the M2 model (Table 3).
Additionally, we examined the effects of the phylogeny on the Pina analysis by utilizing four additional trees. Three of them were generated by removing one at a time, and in combination, the suspected selected sites (i.e., with posterior probability p b ≥ 95%); and the fourth one was constructed based on the Gsp-1 gene genealogy. The ML scores and sites under selection were very similar to the estimates presented in Table 3, suggesting that the inference of sites under positive selection was not affected by the tree topology.
In contrast to Pina, a similar ML analysis for Pinb and Gsp-1 genes revealed no evidence of positive selection (see Supplementary Table S1). The LRTs based on the comparisons of M1a–M2a and M7–M8 did not give statistical support for any of the two datasets. The log-likelihoods for all models were indistinguishable and no codon sites were predicted to be evolving under adaptation. Besides, all ω ratios but one were <1.0; the only exception was an ω ratio >1 under the M8 model for Gsp-1. However, with nonsignificant LRTs, this result may be explained by chance effects, particularly if some codon sites in the sequence, like those observed in Gsp-1, evidence relaxed selective constraint with an underlying ω less than but close to 1 (Anisimova et al. 2002). Conversely, a high number of sites along the Pinb sequences were indicated to be under purifying selection (∼ 85% with ω < 1) compared to Gsp-1 (∼ 60%).
Furthermore, given the complex Pinb phylogeny, which consisted of orthologous/paralogous sequences, we ran the site-specific models with alternative tree topologies using different data sets, including the Triticum/Aegilops lineage alone. Nevertheless, neither of the tests of positive selection (LRTs) nor of the prediction of positively selected sites (BEB) in the Pinb analyses yielded statistically significant results (results not shown).
We further tested for intragenic recombination at individual gene loci. The maximum chi-square method (Maynard Smith 1992) did not detect significant breakpoints (with p > 0.05) in any of the sequence comparisons (results not shown). Likewise, Sawyer’s runs test using GENECONV found no statistical evidence to support recombination events.
Discussion
This study identified and characterized the entire coding region of Pina, Pinb, and Gsp-1 from rye, barley, and all the genomes of the diploid Triticum/Aegilops taxa to elucidate phylogenetic relationships as the bases for testing hypotheses on gene sequence evolution. Phylogenetic comparisons with estimates of evolutionary rates provided insights into the evolutionary dynamics that shaped sequence variation at the Hardness locus-related genes and identified target DNA sequences and amino acid residues that will be valuable for future functional experiments.
Positive diversifying selection appeared to drive the evolution of at least one of the three genes examined, suggesting that adaptive forces have played a role in the preservation and potential functional innovations of duplicate genes at the Hardness locus of the Triticeae genomes. The ML analysis using the d N /d S ratio from interspecific data revealed a strong signature of positive selection (ω > 4) at Pina and detected certain amino acid residues along the mature PIN-a protein with a high probability (>95%) of having evolved under adaptation. These findings from interspecific sequence comparisons further suggested that selection occurred in early stages of Pina evolution. Since no recent action of positive selection has been detected at this locus (Massa et al. 2004), it is likely that fixation of adaptive mutations occurred at the time of speciation.
The pattern of Pina evolution is consistent with the hypothesis that plant defense-related genes are subjected to nonneutral evolution (Parniske et al. 1997; Meyers et al. 1998; Wang et al. 1998; Zhang et al. 2002), and it is also consistent with the observation that evolutionary dynamics of antimicrobial peptides are governed by adaptive forces (Silverstein et al. 2005; Tennessen 2005). Although there is no direct evidence that PIN-a protein alone actually has contributed in vivo to the innate immune system of wheat or any other member of the tribe Triticeae, it is tempting to hypothesize that Pina has evolved adaptively in response to plant pathogens to enhance fitness.
In contrast to Pina, the results for Pinb evidenced no action of positive selection regardless of whether we included the complete or partial data sets in our analyses. These results do not allow us to assert the absence of positive selection (Wong et al. 2004; Zhang et al. 2005). However, it is quite possible that the closely related Pina and Pinb genes (70% amino acid similarity), which share overlapping functions, evolved under different selective constraints associated with variation in their defense function in vivo. It has been shown that closely related antimicrobial peptides (paralogous loci) with different selective patterns also exhibit variation in their antimicrobial properties. Thus, for example, the adaptively evolving gene has improved antimicrobial activity compared to that evolving neutrally or under weak selection (Nicolas et al. 2003; Tennessen 2005). Consistent with this model, the 13-residue fragment of wheat PIN-a (FPVTWRWWKWWKG-NH2; puroA), which has been proposed as the bactericidal domain of PIN-a, has higher in vitro antimicrobial activity than the corresponding peptide of PIN-b (FPVTWPTKWWKG-NH2; puroB) (Jing et al. 2003). The larger number of tryptophan and positively charged amino acid residues that characterize puroA may enhance its antimicrobial properties (Jing et al. 2003). It is interesting that all S-genome Aegilops species analyzed in this study have a WWR instead of a WWK sequence. Arginine (R) and lysine (K) both can interact with tryptophan through cation-π interactions, but based on statistical analysis, the occurrence of arginine is most likely (Gallivan and Dougherty 1999). Cation-π interactions are thought to contribute to protein stability, however, the effect of having an R instead of a K on the antimicrobial properties of PIN-a needs to be investigated.
Our results for Gsp-1 showed some evidence of relaxation of functional constraints with no radical selective pressures, suggesting that this gene is evolving neutrally. Since no apparent selection has altered its evolutionary rates, the Gsp-1 gene is a good candidate for studying the evolution of the tribe Triticeae. The specific biological function of Gsp-1 remains unknown and is still a subject of continued research. Experimental work has demonstrated that Gsp-1 has no significant effect on grain endosperm texture (Tranquilli et al. 2002), but a possible role as a plant defense gene has not been investigated.
This study performed interspecific DNA sequence comparisons for identifying the selective forces associated with gene duplication and provided insights into the macroevolutionary dynamics of PINs and Gsp-1 genes in the tribe Triticeae. Results suggested the role of positive selection during the evolution of the Hardness locus-related genes and supported previous studies (Silverstein et al. 2005; Tennessen 2005), suggesting that fixation of adaptive mutations by natural selection might operate in the evolution of antimicrobial peptides. Additionally, we documented novel PIN genomic sequences in the wild wheat germ plasm, rye, and barley that will be useful for the assessment of grain texture in wheat and other cereal crops.
References
Anisimova M, Bielawski JP, Yang Z. (2002) Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol 19:950–958
Beecher B, Smidansky ED, See D, Blake TK, Giroux MJ (2001) Mapping and sequence analysis of barley puroindolines. Theor Appl Genet 102:833–840
Caldwell KS, Langridge P, Powell W (2004) Comparative sequence analysis of the region harboring the hardness locus in barley and its collinear region in rice. Plant Physiol 136:3177–3190
Chantret N, Cenci A, Sabot F, Anderson OD, Dubcovsky J (2004) Sequencing of the Triticum monococcum hardness locus reveals good microcolinearity with rice. Mol Genet Genomics 271:377–386
Chantret N, Salse J, Sabot F, Rahman S, Bellec A, Laubin B, Dubois I, Dossat C, Sourdille P, Joudrier P, Gautier MF, Cattolico L, Beckert M, Aubourg S, Weissenbach J, Caboche M, Bernard M, Leroy P, Chalhoub B (2005) Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploid wheat species (Triticum and Aegilops). Plant Cell 17:1033–1045
Clark AG (1994) Invasion and maintenance of a gene duplication. Proc Natl Acad Sci USA 91:2950–2954
Darlington HF, Rouster J, Hoffmann L, Halford NG, Shewry PR, Simpson DJ (2001) Identification and molecular characterization of hordoindolines from barley grain. Plant Mol Biol 47:785–794
Dubreil L, Gaborit T, Bouchet B, Gallant DJ, Broekaert WF, Quillien L, Marion D (1998) Spatial and temporal distribution of the major isoforms of puroindolines (puroindoline-a and puroindoline-b) and non-specific lipid transfer protein (ns-LTPe1) of Triticum aestivum seeds. Relationships with their in vivo antifungal properties. Plant Sci 138:121–135
Endo TR, Gill BS (1996) The deletion stocks of common wheat. J Hered 87:295–307
Gallivan JP, Dougherty DA (1999) Cation-π-interactions in structural biology. Proc Natl Acad Sci USA 96:9459–9464
Gautier MF, Cosson P, Guirao A, Alary R, Joudrier P (2000) Puroindoline genes are highly conserved in diploid ancestor wheat and related species but absent in tetraploid Triticum species. Plant Sci 153:81–91
Giroux MJ, Morris CF (1997) A glycine to serine change in puroindoline b is associated with wheat grain hardness and low levels of starch-surface friabilin. Theor Appl Genet 95:857–864
Giroux MJ, Morris CF (1998) Wheat grain hardness results from highly conserved mutations in the friabilin components puroindoline a and b. Proc Natl Acad Sci USA 95:6262–6266
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jing W, Demcoe AR, Vogel HJ (2003) Conformation of a bactericidal domain of puroindoline a: structure and mechanism of action of a 13-residue antimicrobial peptide. J Bacteriol 185:4938–4947
Krishnamurthy K, Balconi C, Sherwood JE, Giroux MJ (2001) Wheat puroindoline enhance fungal disease resistance in transgenic rice. MPMI 14:1255-1260
Le Guerneve C, Seigneuret M, Marion D (1998) Interaction of the wheat endosperm lipid-binding protein puroindoline-a with phospholipids. Arch Biochem Biophys 360:179–186
Li WH, Wu CI, Luo CC (1985) A new method for estimating synonymous and nonsynonymous rates of nucleotide substitutions considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol 2:150–174
Lillemo M, Simeone MC, Morris CF (2002) Analysis of puroindoline a and b sequences from Triticum aestivum cv. ‘Penawawa’ and related diploid taxa. Euphytica 126:321–331
Lynch M, O’Hely M, Walsh B, Force A (2001) The probability of fixation of a newly arisen gene duplicate. Genetics 159:1789–1804
Massa AN, Morris CF, Gill BS (2004) Sequence diversity of puroindoline-a, puroindoline-b, and the grain softness protein genes in Aegilops tauschii Coss. Crop Sci 44:1808–1816
Maynard Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34:126–129
Meyers BC, Shen KA, Rohani P, Gaut BS, Michelmore RW (1998) Receptor-like genes in the major resistance locus of lettuce are subject to divergent selection. Plant Cell 10:1833–1846
Miyata T, Miyazawa S, Yasunaga T (1979) Two types of amino acid substitutions in protein evolution. J Mol Evol 12:219–236
Moore RC, Purugganan MD (2003) The early stages of duplicate gene evolution. Proc Natl Acad Sci USA 100:15682–15687
Morris CF 2002. Puroindolines: The molecular genetic basis of wheat hardness. Plant Mol Biol 48:633–647
Nicolas P, Vanhoye D, Amiche M (2003) Molecular strategies in biological evolution of antimicrobial peptides. Peptides 24:1669–1680
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Ohno S (1970) Evolution by gene duplication. Springer Verlag, New York
Ozkan H, Levy A A, Feldman M (2001) Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell 13: 1735–1747
Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, Wulff BBH, Jones JDG (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821–832
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:917–818
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Sawyer S. (1989) Statistical tests for detecting gene conversion. Mol Biol Evol 6:526–538
Shewry PR, Morell M (2001) Manipulating cereal endosperm structure, development and composition to improve end-use properties. Adv Bot Res 34:165–236
Silverstein KAT, Graham MA, Paape TD, VandenBosch KA (2005) Genome organization of more than 300 defensin-like genes in Arabidopsis. Plant Physiol 138:600–610
van Slageren MW (1994) Wild wheats: a monograph of Aegilops L. and Ambylopyrum (Jaub. & Spach) Eig (Poaceae). Wageningen Agricultural University and ICARDA, Wageningen, the Netherlands
Swofford DL (2002) PAUP: Phylogenetic Analysis Using Parsimony, version 4.0b 10. Sinauer Associates, Sunderland, MA
Tennessen JA (2005) Molecular evolution of animal antimicrobial peptides: widespread moderate positive selection. J Evol Biol 18:1387–1394
Thompson JD, Higgins DG, Gibson TJ (1994) ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tranquilli G, Lijavetzky D, Muzzi G, Dubcovsky J (1999) Genetic and physical characterization of grain texture-related loci in diploid wheat. Mol Gen Genet 262:846–850
Tranquilli G, Heaton J, Chicaiza O, Dubcovsky J (2002) Substitutions and deletions of genes related to grain hardness in wheat and their effect on grain texture. Crop Sci 42:1812–1817
Wang GL, Ruan DL, Song WY, Sideris S, Chen L, Pi LY, Zhang S, Zhang Z, Fauquet C, Gaut B S, Whalen MC, Ronald PC (1998) Xa21D encodes a receptor-like molecule with a leucine-rich repeat domain that determines race-specific recognition and is subject to adaptive evolution. Plant Cell 10:765–780
Wong WSW, Yang Z, Goldman N, Nielsen R (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 15:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AMK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107-1118
Zhang L, Peek AS, Dunams D, Gaut BS (2002) Population genetics of duplicated disease-defense genes, hm1 and hm2 in maize (Zea mays ssp. mays L.) and its wild ancestors (Zea mays ssp. parviglumis). Genetics 162:851–860
Acknowledgments
We thank Dr. Andris Kleinhofs for providing seeds of barley cv. Morex and Dr. Philip Greenwell for constructive comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Magnus Nordborg]
Mention of trademark or proprietary products does not constitute a guarantee or warranty of a product by the U.S. Department of Agriculture and does not imply its approval to the exclusion of other products that may also be suitable. This article is in the public domain and not copyrightable.
An erratum to this article is available at http://dx.doi.org/10.1007/s00239-006-8292-1.
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Massa, A.N., Morris, C.F. Molecular Evolution of the Puroindoline-a, Puroindoline-b, and Grain Softness Protein-1 Genes in the Tribe Triticeae. J Mol Evol 63, 526–536 (2006). https://doi.org/10.1007/s00239-005-0292-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0292-z