
Abstract
Sequence directed mutagenesis is a mechanism by which imperfect repeats “repair” each other to become perfect, generating mutations. This process is known to be prevalent in prokaryotes and it has been implicated in several human genetic diseases. Here we test whether sequence directed mutagenesis occurs in the protein coding sequences of eukaryotes using extensive DNA sequence data from humans, mice, Drosophila, nematodes, yeast, and Arabidopsis. Using two tests we find little evidence of sequence directed mutagenesis. We conclude that sequence directed mutagenesis is not prevalent in eukaryotes and that the examples of human diseases, apparently caused by sequence directed mutagenesis, are probably coincidental.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
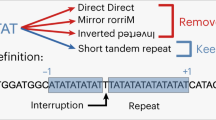
The genomes of most organisms are littered with short inverted repeats which often differ from one another by a few nucleotides. These are potentially a source of mutations because imperfect repeats can undergo a process known as sequence directed mutagenesis, which generates perfect repeats and a set of mutations. The process by which this occurs is not fully understood. It seems to involve template switching and the formation of hairpin structure during DNA replication (Ripley 1982), which facilitates the “correction” of the imperfect repeats. Sequence directed mutagenesis does not appear to involve recombination and subsequently is different from gene conversion. The process has been directly demonstrated in a number of bacteria and a recent survey of bacterial genomes showed that almost all have an excess of short (7-bp) inverted repeats, suggesting that sequence directed mutagenesis is common in both eubacterial and archeabacterial genomes (van Noort et al. 2003). Furthermore, computer simulations have shown that sequence directed mutagenesis might be an important potential mutagenic source (Fieldhouse and Golding 1991).
The process has been less extensively studied in eukaryotes. Concurrent substitutions in human interferon (Golding and Glickman 1985), frameshift mutations in yeast (Ripley 1982), and a list of human diseases (Blisser 1998) have been attributed to sequence directed mutagenesis. But the role or frequency of sequence directed mutagenesis has never been firmly established. A recent analysis has shown that eukaryotic genomes have an excess of inverted repeats (Cox and Mirkin 1997), but many of these appear to be due to recent duplications (Thomas et al. 2004).
Here we set out to test whether sequence directed mutagenesis occurs in eukaryotic genomes using two tests. First, we test whether inverted repeats are more common than would be expected by chance. Second, we investigate whether perfect inverted repeats tend to be close together.
Materials and Methods
To perform our analysis we extracted all protein coding exons which were longer than 2000 bp from Caenorhabditis elegans, Drosophila melanogaster, Mus musculus, Homo sapiens, Saccharomyces cerevisiae, and Arabidopsis thaliana genomes using the ACNUC retrieval system. We restricted our analysis to exons for two reasons. First, we were initially interested in whether sequence directed mutagenesis was generating mutations associated with human disease, and most mutations which cause Mendelian diseases are found in protein coding sequences (Stenson et al. 2003). Second, considering coding sequences reduces the problem of repeats generated by transposable elements and spontaneous duplications of small sequences (Thomas et al. 2004). We restricted our analysis to exons of >2000 bp to ensure proper randomization of the sequences in our analysis. In total we retrieved 139 exons from C. elegans, 742 for Drosophila, 492 for human, 404 for mouse, 1209 for yeast, and 1039 for Arabidopsis.
To identify repeats we slid a window of 50, l00, and 500 bp across each exon. Within that window we counted (a) the number of perfect inverted repeats 6, 7, 8, and 9 bp long and (b) the distance between those repeats. Repeats of a certain length contained only the repeats of that length, not repeats which were shorter. For example, the number of 7-bp repeats did not contain the number of 6-bp repeats because those were of a different class.
The numbers of repeats and distances were also counted in randomized sequences. To randomize the sequence we swapped synonymous codons, thus preserving the amino acid sequence and codon usage of the sequence. Because nucleotides can affect the mutation pattern of their adjacent neighbors, we further restricted the randomization by only swapping synonymous codons which were followed by the same base (e.g., CAA.G could be swapped with CAT.G but not CAT.C). Because this randomization scheme greatly reduces the number of permutations that are possible, we restricted our analysis to exons which were >2000 bp in length. In such a sequence each sense codon, followed by each nucleotide, is represented on average 2.7 times, which means that a twofold degenerate amino acid will have on average 5.4 codons, and a fourfold degenerate amino acid 10.8 codons––i.e., we expect most codons to be represented more than once and randomization to be possible. Analyses with exons >4000 bp in length gave qualitatively similar results.
van Noort et al. (2003) adopted a different strategy to assess whether perfect repeats were more common than expected by chance; they compared the relative number of perfect repeats to the relative number of imperfect repeats, which differed from one another by one nucleotide, where the number of repeats in each case was relative to the number expected from the dinucleotide frequencies. However, this approach is only valid if dinucleotide frequencies are homogeneous across the genome, which they are not in many organisms. For example, mammals shows very strong heterogeneity in base composition (Bernardi 1993; Lander et al. 2001; Waterston et al. 2002).
Results and Discussion
If sequence directed mutagenesis is occurring, we would expect perfect repeats to be more common than by chance. To test this we counted the number of perfect repeats of between 6 and 9 bp in a window of 50, 100, and 500 bp and compared this to the number we found when each exon was randomized.
In our study human, mice, and Arabidopsis show significantly more repeats than expected by chance, whereas Drosophila shows a significant deficit (Fig. 1). Similar patterns are observed for the repeat classes individually and for all window sizes. However, although human, mice and Arabidopsis show significant excess, the size of the excess is very small; we estimate that the repeats are <5% more common than expected by chance. It is therefore apparent that if sequence directed mutagenesis is occurring in humans and mice, it is a weak force.

Ratio of the observed to the expected number of perfect inverted repeats in five eukaryotic genomes. Bars indicate standard errors. CE: C. elegans, DM: Drosophila, HS: humans, MM: mouse, SC: yeast, AT: Arabidopsis.
We would also expect, given current models of sequence directed mutagenesis, which involve template switching during DNA replication (Ripley 1982), to find perfect repeats clustered together. In all species there is an excess of two perfect inverted repeats which are adjacent––but otherwise there is no indication that inverted repeats tend to lie close to each other (Fig. 2). It seems unlikely that the adjacent perfect repeats are a consequence of sequence directed mutagenesis since all models of sequence directed mutagenesis require the DNA to bend between the two repeats and several of our species show evidence of these adjacent perfect repeats but no evidence of an excess of repeats over random expectations––for example, Drosophila.

Number of perfect inverted repeats as a function of the distance (bp) between the two repeat copies. The genomes of C. elegans (CE), Drosophila (DM), human (HS), mouse (MM), yeast (SC), and Arabidopsis (AT) are represented by diamonds, squares, triangles, X’s, crosses, and filled circles, respectively.
Although there is a significant excess of perfect inverted repeats in both of the mammals we have studied and in Arabidopsis, the excess is quite small, which suggests either that sequence directed mutagenesis does not occur or that it occurs at a low frequency. This perspective is corroborated by the fact that perfect repeats are not clustered unless they form perfect palindromes. The slight excess of perfect repeats may be due to neighboring base effects which extend beyond the adjacent nucleotide. The lack of any strong evidence of sequence directed mutagenesis is in sharp contrast to bacterial genomes, where there is a marked excess of perfect repeats (van Noort et al. 2003). The absence or low rate of sequence directed mutagenesis in the human genome raises the question why some human diseases appear to be caused by sequence directed mutagenesis, for example, antithrombin deficiency and osteogenesis imperfecta (Blisser 1998). We would argue that these are coincidences. Every genome contains a huge number of perfect repeats and an even larger number of imperfect repeats, so it is quite likely that several of the 25,000 mutations associated with Mendelian diseases in humans (Stenson et al. 2003) will be close to an imperfect repeat, which could generate the disease mutation.
References
Bernardi G (1993) The isochore organization of the human genome and its evolutionary history––a review. Gene 135:57–66
Blisser JJ (1998) DNA inverted repeats and human diseases. Front Biosci 3:d408–418
Cox R, Mirkin SM (1997) Characteristic enrichment of DNA repeats in different genomes. Proc Natl Acad Sci USA 94:5237–5242
Fieldhouse D, Golding B (1991) A source of small repeats in genomic DNA. Genetics 129:563–572
Golding GB, Glickman BW (1985) Sequence-directed mutagenesis: evidence from a phylogenetic history of human alpha-interferon genes. Proc Natl Acad Sci USA 82:8577–8581
Lander ES, Linton LM, Birren B, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Ripley LS (1982) Model for the participation of quasi-palindromic DNA sequences in frameshift mutation. Proc Natl Acad Sci USA 79:4128–4132
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN (2003) Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21:577–581
Thomas EE, Srebro N, Sebat J, Navin N, Healy J, Mishra B, Wigler M (2004) Distribution of short paired duplications in mammalian genomes. Proc Natl Acad Sci USA 101:10349–10354
van Noort V, Worning P, Ussery DW, Rosche WA, Sinden RR (2003) Strand misalignments lead to quasipalindrome correction. Trends Genet 19:365–369
Waterston RH, Lindblad-Toh K, Birney E, et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Acknowledgments
We thank Laurent Duret for providing the sequences. This project was funded by EU, Marie Curie Fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Richard Kliman]
Rights and permissions
About this article
Cite this article
Ladoukakis, E.D., Eyre-Walker, A. Searching for Sequence Directed Mutagenesis in Eukaryotes. J Mol Evol 64, 1–3 (2007). https://doi.org/10.1007/s00239-005-0120-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0120-5