
Abstract
Following cessation of recombination during sex chromosome evolution, the nonrecombining sex chromosome is affected by a number of degenerative forces, possibly resulting in the fixation of deleterious mutations. This might take place because of weak selection against recessive or partly recessive deleterious mutations due to permanent heterozygosity of nonrecombining chromosomes. Furthermore, population genetic processes, such as selective sweeps, background selection, and Muller’s ratchet, result in a reduction in Ne, which increase the likelihood of fixation of deleterious mutations. Theory thus predicts that nonrecombining genes should show increased levels of nonsynonymous (dN) to synonymous substitutions (dS). We tested this in an avian system by estimating the ratio between dN and dS in six gametologous gene pairs located on the Z chromosome and the nonrecombining, female-specific W chromosome. In comparisons, we found a significantly higher dN/dS ratio for the W-linked than the Z-linked copy in three of the investigated genes. In a concatenated alignment of all six genes, the dN/dS ratio was six times higher for W-linked than Z-linked genes. By using human and mouse as outgroup in maximum likelihood analyses, W-linked genes were found to evolve differently compared with their Z-linked gametologues and outgroup sequences. This seems not to be a consequence of functional diversification because dN/dS ratios between gametologous gene copies were consistently low. We conclude that deleterious mutations are accumulating at a high rate on the avian W chromosome, probably as a result of the lack of recombination in this female-specific chromosome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Models of sex chromosome evolution postulate that selection for linkage between a sex-determining locus and sexually antagonistic alleles will favor suppression of recombination between the proto-sex chromosomes (Charlesworth 1978; Charlesworth and Charlesworth 2000; Rice 1996). Once initiated, this process is expected to lead to the differentiation of the two sex chromosomes, and the concurrent degeneration of the nonrecombining chromosome. Theory suggests that a nonrecombining chromosome will be subject to a number of degenerative forces (Charlesworth and Charlesworth 2000). Nonrecombining chromosomes should be prone to accumulate deleterious mutations because natural selection is less effective when a gene is always present in heterozygote state (Muller 1918). In addition, deleterious mutations are expected to accumulate due to stochastic processes such as Muller’s ratchet (the loss of the class of chromosomes least loaded with mutations) (Felsenstein 1974), by hitchhiking arising from selective sweeps of advantageous mutations (Rice 1987) and due to background selection (Charlesworth 1994).
While sex chromosome evolution and chromosome degeneration following from lack of recombination have been well studied theoretically, empirical data from molecular evolutionary analyses have started to accumulate only recently. Four groups of organisms have been used for this research: mammals (Agulnik et al. 1997; Wyckoff et al. 2002; Tucker et al. 2003), Drosophila (Bachtrog and Charlesworth 2002), plants of the genus Silene (Filatov and Charlesworth 2002), and birds (Fridolfsson and Ellegren 2000). Comparative studies of sex chromosome evolution are important because although sex chromosomes are found in a diverse range of eukaryotes, comparative mapping suggests that sex chromosomes have evolved independently many times during evolution. Equally important, sex chromosome evolution has taken place in quite different genetic systems and over different time scales. For instance, mammalian sex chromosome evolution is estimated to have been initiated 320 million years ago (MYA) (Lahn and Page 1999), which is much earlier than the evolution of the neo-sex chromosomes of Drosophila miranda (1.25 MYA [Yi and Charlesworth 2000]) and Silene (<10 MYA [Nicolas et al. 2005]). Birds, in which sex chromosome evolution was initiated 100–170 MYA (Handley et al. 2004), are intermediate. Moreover, the genetic mechanism of sex determination is not uniform among organisms with sex chromosomes; the Y chromosome has a dominant role in mammals, whereas in flies the X chromosome has a dosage effect. Furthermore, mammals, flies and plants represent sex chromosome systems with male heterogamety (males, XY; females, XX), where degenerative forces acting on the nonrecombining Y chromosome. Y chromosome degeneration may be augmented or counteracted by the forces of sexual selection for male-specific Y-linked traits. On the other hand, in birds, females are the heterogametic sex (males, ZZ; females, ZW). Contrasting the patterns of sex chromosome evolution in different genetic systems may therefore be useful for understanding processes ultimately associated with sex chromosome evolution and degeneration of nonrecombining chromosomes.
After sufficient time has elapsed since their emergence, nonrecombining sex chromosomes like the mammalian Y chromosome (Skaletsky et al. 2003) and the avian W chromosome (Chicken Genome Consortium 2004) are often small and lack many of the genes present on X/Z. These observations alone can be seen as the result of sex chromosome degeneration following suppressed recombination. But to investigate ongoing processes of genetic degeneration under the assumption that the efficacy of selection is reduced in nonrecombining regions is less straightforward when sex chromosomes are ancient and where few signs of these processes may remain. However, one possible approach is offered by the analysis of rates of nucleotide substitution on sex chromosomes. Specifically, theory predicts that deleterious mutations, manifested by nonsynonymous nucleotide substitutions, should accumulate at a higher rate on the nonrecombining Y/W chromosome than on X/Z (Charlesworth 1996). Since the two sex chromosomes may have different overall rates of mutation (Hurst and Ellegren 1998; Li et al. 2002), the rate of accumulation of deleterious mutation can be compared by taking the nonsynonymous (dN)-to-synonymous (dS) substitution rate ratio (dN/dS). The amounts of selective constraint, and hence dN/dS ratios, clearly vary among genes, so large sample sizes are needed to obtain representative estimates of the mean dN/dS ratio for individual chromosomes. Alternatively, and more feasibly, comparison of dN/dS ratios in genes shared between the two sex chromosomes (gametologous genes, or gametologues) allows one to study the same gene exposed to different genomic environment (Wyckoff et al. 2002). Of course, this assumes that gene divergence has not been accompanied by functional diversification, which could alter selection regimes. Here we seek to gain further insight into sex chromosome evolution by studying accumulation of deleterious mutations on the female-specific W chromosome of birds. We use six gametologous gene pairs on the Z and W sex chromosomes and estimate rates of divergence between chicken (Gallus gallus) and turkey (Meleagris gallopavo) or zebra finch (Taeniopygia guttata).
Materials and Methods
Genes and Species
Chicken (Gallus gallus) and turkey (Meleagris gallopavo) sequences for parts of the gametologous gene pairs MADH2Z/MADH2W, SPINZ/SPINW, UBAP2Z/UBAP2W, IDN3Z/IDN3W, and RASA1Z/RASA1W were either taken from databases or obtained by sequencing. Chicken and zebra finch (Taeniopygia guttata) sequences for CHD1Z/CHD1W were taken from databases. The following sequences were collected from GenBank: chicken CHD1Z (AF004397) and CHD1W (AF181826), zebra finch CHD1Z (AY217131) and CHD1W (AY217129), chicken MADH2Z (NM_204561), chicken SPINZ (AB047853) and SPINW (AB047852), chicken UBAP2Z (AY188762) and UBAP2W (AY188763), turkey UBAP2W (AY188758). Chicken IDN3Z, IDN3W, MADH2W, RASA1Z, and RASA1W were from the draft assembly of the chicken genome available at the Web site http://www.ensembl.org/. Human and mouse (Mus musculus) orthologues were taken from GenBank: CHD1 (NM_001270, L10410), IDN3 (NM_133433, NM_027707), MADH2 (U68018, BC021342), RASA1 (NM_022650, NM_145452), SPIN (BT007314, BC016517), and UBAP2 (NM_018449, NM_026872). Chicken and turkey belong to the order Galliformes, and zebra finch to the order Passeriformes. The time of divergence between chicken and turkey has been estimated to be 28–45 MYA (Dimcheff et al 2002; van Tuinen and Dyke 2004), while the time of divergence between Galliformes and Passeriformes is roughly 80–100 MYA ( van Tuinen and Hedges 2001; van Tuinen and Dyke 2004).
PCR Amplification and DNA Sequencing
Parts of IDN3Z, IDN3W, MADH2Z, MADH2W, RASA1Z, RASA1W, SPINZ, SPINW, and UBAP2Z were sequenced in turkey (GenBank accession numbers DQ090090–DQ090122). We amplified coding sequence from genomic DNA using Z or W chromosome-specific primers designed from intron sequences in the chicken genome assembly (supplementary information). PCRs were performed in 20-μl volumes on a Perkin Elmer 9600 Thermal Cycler using 0.5 U of AmpliTaq Gold, 2.5 mM MgCl2, 0.08 mM dNTPs, 1× PCR Gold Buffer (Applied Biosystems), 5 pmol of each primer, and 50 ng of template DNA. PCR products were separated on 2% agarose gels, run in 0.5% TAE buffer, and visualized by ethidium bromide staining. PCR-products were purified with ExoSAP-IT reagent (Amersham Biosciences), followed by direct sequencing in forward and reverse directions using the DYEnamic ET DyeTerminator Kit (Amersham Biosciences). Sequencing reactions were electrophoresed on a MegaBACE 1000 sequencing instrument (Amersham Bioscences). Z-linked genes were amplified from male DNA, while female DNA was used as template for amplification of W-linked genes. In order to confirm Z- or W-linkage of the turkey sequences obtained, phylogenetic analysis with both gametologues from chicken was performed (data not shown).
Sequence Analysis
Sequences were edited in Autoassembler (Applied Biosystems) and overlapping forward and reverse sequences were compared to construct consensus sequences. After translation into amino acid sequence, alignments were made using Clustal W. We used MEGA 3.0 (Kumar et al. 2004) to estimate numbers of nonsynonymous substitutions (N) and numbers of synonymous substitutions (S), applying the Nei-Gojobori (1986) method and Jukes-Cantor (1969) model to correct for multiple hits. In order to statistically test if N and S were different for Z- and W-linked genes, we used substitutions in concatenated alignments of all genes, for all genes except CHD1, as well as for each gene, and performed chi-square tests (analogous to Wyckoff et al. 2002). Maximum likelihood analyses were conducted using codeml in the PAML 3.14a package (Yang 1997), allowing for heterogeneity in dN/dS (this ratio denoted ω) among lineages in a phylogeny (Goldman and Yang 1994; Yang 1998). These analyses were performed using a concatenated dataset, as well as a concatenated dataset of all genes in the chicken-turkey comparison (CHD1 excluded). The simplest model assumes that the same ω ratio holds for all lineages in the phylogeny and is referred to as the one-ratio model. The most liberal model is the free-ratio model, which contains as many ω-parameters as branches in the phylogeny. We also included two intermediate models: a two-ratio model, where the ω ratio was allowed to vary between W chromosome lineages and Z chromosome and outgroup (human and mouse) lineages; and a three-ratio model, where ω varied between W chromosome, Z chromosome, and outgroups. Given the data and the retrieved phylogeny, the fit of these four models was statistically compared using likelihood ratio tests. These tests compares twice the difference in log likelihood between two models to a chi-square distribution with the number of degrees of freedom equal to the difference in free parameters between the models.
Results
Orthologous sequences were obtained from chicken and turkey for five gametologous gene pairs on the Z and W sex chromosomes: MADH2Z/MADH2W (417/390 codons), SPINZ/SPINW (238/249), UBAP2Z/UBAP2W (150/188), IDN3Z/IDN3W (276/281), and RASA1Z/RASA1W (198/178), For a sixth gene pair, CHD1Z/CHD1W, orthologous sequences from chicken and zebra finch (980/980 codons) were used. Rates of synonymous substitution in the chicken-turkey and chicken-zebra finch comparisons are uniformly higher for Z-linked than for W-linked genes (Table 1). This is in agreement with previous findings, mainly based on intronic sequence divergence, of a male-biased avian mutation rate (e.g., Ellegren and Fridolfsson 1997). The male-to-female mutation rate ratio (αm) estimated according to Miyata et al. (1987), using dS in a concatenated alignment (Z, 1275 codons; W, 1282 codons) of all five genes in the chicken-turkey comparison (i.e., CHD1 excluded), is similar (2.80) to the estimate obtained in a recent analysis of 43 kb noncoding autosomal, Z-linked and W-linked DNA in chicken and turkey (2.47; Axelsson et al. 2004).
Between species, the pairwise dN/dS ratio is significantly higher for SPINW than for SPINZ (p = 0.01), for UBAP2W than for UBAP2Z (p = 0.03) and for CHD1W than for CHD1Z (p < 0.0001; Table 1). For all the other gene pairs the dN/dS ratio was higher for W than Z, but the difference was not significant; however, very few nonsynonymous substitutions are observed in these genes. For the concatenated alignment of all six genes (Z, 2267 codons; W, 2275), dN/dS is about six times higher for W-linked (0.080) than for Z-linked gametologues (0.014; p < 0.0001). With a concatenated alignment without CHD1, dN/dS is approximately seven times higher for W-linked (0.089) than Z-linked genes (0.012; p < 0.001). It thus seems that deleterious mutations generally accumulate at a higher rate on the avian W chromosome than on the Z chromosome.
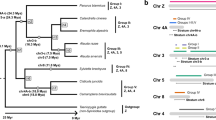
Parameters estimated in the maximum likelihood analyses, in which human and mouse outgroup sequences were included, clearly indicate an excess of nonsynonymous substitutions in W chromosome lineages compared to Z chromosome lineages (Fig. 1). For both the six-gene and the five-gene dataset (CHD1 excluded), likelihood ratio tests show that the two-ratio as well as the three-ratio and free-ratio models all represent a significantly better fit to the data than the one-ratio model (p < 0.0001 in all cases; Table 2). Moreover, the three- and free-ratio models are not significantly better than the two-ratio model. Taken together, these analyses suggest that the two-ratio model best describes the data and indicate that these W-linked genes evolve in a manner different from their Z-linked gametologues and orthologous sequences in human and mouse.

Maximum likelihood parameters estimated under the free ratio model (concatenated six-gene dataset).
Discussion
Our data demonstrate that deleterious mutations accumulate at a higher rate on the W than on the Z chromosome in birds. How do our observations relate to what have been seen in other organisms? Only a handful of gametologous gene pairs have been investigated for dN/dS ratios on the mammalian X and Y chromosomes (Agulnik et al. 1997; Wyckoff et al. 2002), and the same applies to the neo-sex chromosomes of Drosophila miranda (Yi and Charlesworth 2000; Bachtrog and Charlesworth 2002; Bachtrog 2003) and the sex chromosomes of Silene (Filatov and Charlesworth 2002; Filatov 2005; Nicolas et al. 2005). A higher dN/dS ratio for the Y-linked than the X-linked gametologue has been found for most, but not all, of these genes. With the data presented here for birds, the overall pattern in the four groups of organisms is thus one of elevated rates of accumulation of nonsynonymous substitutions on the nonrecombining sex chromosome. Moreover, the trend is seen in genetic systems of male as well as female heterogamety. Independent analyses in different groups of organisms is important in studies of general features of sex chromosome evolution, because even if several genes from one particular system show a similar evolutionary pattern, they are all part of the same evolutionary trajectory and thus essentially correspond to a single observation.
Ideally, incipient W/Y chromosomes should be studied for the degenerative effects of suppressed recombination, as long-term divergence between gametologues may lead to elevated dN/dS ratios of W-/Y-linked genes due to divergence in function, driven by positive selection, rather than lower efficacy of selection ( Gerrad and Filatov 2005).However, low dN (<0.02) and dN/dS ratios (<0.1) between the Z- and the W-linked gametologues of all six gene pairs studied suggest lack of functional diversification in W-linked genes (Table 3). Furthermore, in comparison to another gametologous gene pair in birds, HINTZ/HINTW, molecular evolutionary data from CHD1Z/CHD1W, IDN3Z/IDN3W, MADH2Z/MADH2W, RASA1Z/RASA1W, SPINZ/SPINW, and UBAP2Z/UBAP2W indicate that these gametologues have not diverged considerably. In the case of HINTZ/HINTW, in contrast, dN/dS along the branch leading to HINTW is well above 1 with strong evidence of adaptive evolution of a female-specific function (Ceplitis and Ellegren 2004). Of course, this does not exclude the possibility of functionally related differences at individual sites or in regulatory regions of the genes studied here (Itoh et al. 2001; Agate et al. 2004).
The evolutionary consequences of lack of recombination on the avian W chromosome are not only manifested in chromosomal decay and high dN/dS ratios of genes which still remain on the chromosome. Nucleotide diversity on the chicken W chromosome is more than 10-fold lower than neutral expectations (Berlin and Ellegren 2004), and similar observations have been made for other bird species as well (Berlin and Ellegren 2001; Montell et al. 2001). These observations are consistent with a severely reduced effective population size of W caused by selective sweeps and/or background selection. Low levels of nucleotide diversity are also a salient feature of nonrecombining Y chromosomes (Filatov et al. 2000; Sachidanandam et al. 2001; Bachtrog 2004; Hellborg and Ellegren 2004).
According to “the strata hypothesis” (Lahn and Page 1999), cessation of recombination between sex chromosomes takes place in a stepwise fashion and evidence from the avian Z and W chromosomes suggests that this has happened at least two times, creating two evolutionary strata along the Z chromosome (Handley et al. 2004). It has been estimated that recombination in the older stratum 1 ceased at about 100–170 MYA, while recombination continued in stratum 2 until 58–85 MYA (Handley et al. 2004). The genes studied here come from both strata (Table 3), suggesting that the degeneration of W-linked genes is an ongoing process despite very long time of divergence between the Z and the W. Furthermore, there are other signs of ongoing degeneration of avian W chromosomes. One W-linked gene, ATP5A1W, whose Z-linked gametologue ATP5A1Z is located in stratum 2, has recently been lost from the W chromosome of parrots ( de Kloet 2001). Moreover, many copies of the ampliconic HINTW gene have evolved into pseudogenes with frameshift and stop codon mutations in different avian lineages (Hori et al. 2000; O’Neill et al. 2000); multiple copies of a nonrecombining gene might be a way of avoiding accumulation of deleterious mutations (Lahn et al. 2001; Skaletsky et al. 2003).
Further studies should be devoted to more confidently disentangle the effects of adaptive and purifying selection influencing the evolution of W-linked genes. Sequence data sets from multiple species will allow the application of maximum likelihood models which incorporate variable selective pressures across the codons (Yang 1998). Moreover, if polymorphism data can be retrieved, it shall be possible to contrast dN/dS ratios for diversity and divergence, and to use allele frequency data to study population genetic processes.
References
Agate RJ, Choe M, Arnold AP (2004) Sex differences in structure and expression of the sex chromosome genes CHD1Z and CHD1W in zebra finches. Mol Biol Evol 21:384–396
Agulnik AI, Bishop CE, Lerner JL, Agulnik SI, Solovyev VV (1997) Analysis of mutation rates in the SMCY/SMCX genes shows that mammalian evolution is male driven. Mamm Genome 8:134–138
Axelsson E, Smith NGC, Sundstrom H, Berlin S, Ellegren H (2004) Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol Biol Evol 21:1538–1547
Bachtrog D (2003) Adaptation shapes patterns of genome evolution on sexual and asexual chromosomes in Drosophila. Nat Genet 34:215–219
Bachtrog D (2004) Evidence that positive selection drives Y-chromosome degeneration in Drosophila miranda. Nat Genet 36:518–522
Bachtrog D, Charlesworth B (2002) Reduced adaptation of a nonrecombining neo-Y chromosome. Nature 416:323–326
Berlin S, Ellegren H (2001) Clonal inheritance of avian mitochondrial DNA. Nature 413:37–38
Berlin S, Ellegren H (2004) Chicken W: a genetically uniform chromosome in a highly variable genome. Proc Natl Acad Sci USA 101:15967–15969
Ceplitis H, Ellegren H (2004) Adaptive molecular evolution of HINTW, a female-specific gene in birds. Mol Biol Evol 21:249–254
Charlesworth B (1978) Model for evolution of Y chromosomes and dosage compensation. Proc Natl Acad Sci U S A 75:5618–5622
Charlesworth B (1994) The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet Res 63:213–227
Charlesworth B (1996) The evolution of chromosomal sex determination and dosage compensation. Curr Biol 6:149–162
Charlesworth B, Charlesworth D (2000) The degeneration of Y chromosomes. Philos Trans R Soc Lond B 355:1563–1572
Chicken Genome Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432:695–716
de Kloet SR (2001) Loss of the gene for the alpha subunit of ATP synthase (ATP5A1) from the W chromosome in the African grey parrot (Psittacus erithacus). J Mol Evol 53:135–143
de Kloet RS, de Kloet SR (2003) Evolution of the spindlin gene in birds: independent cessation of the recombination of sex chromosomes at the spindlin locus in neognathous birds and tinnamous, a palaeognathous avian family. Genetica 119:333–342
Dimcheff DE, Drovetski SV, Mindell DP (2002) Phylogeny of Tetraoninae and other galliform birds using mitochondrial 12S and ND2 genes. Mol Phylogenet Evol 24:203–15
Ellegren H, Fridolfsson AK (1997) Male-driven evolution of DNA sequences in birds. Nat Genet 17:182–184
Felsenstein J (1974) The evolutionary advantage of recombination. Genetics 78:737–756
Filatov DA (2005) Substitution rates in a new Silene latifolia sex-linked gene, SlssX/Y. Mol Biol Evol 22(3):402–408
Filatov DA, Charlesworth D (2002) Substitution rates in the X- and Y-linked genes of the plants, Silene latifolia and S. dioica. Mol Biol Evol 19:898–907
Filatov DA, Moneger F, Negrutiu I, Charlesworth D (2000) Low variability in a Y-linked plant gene and its implications for Y-chromosome evolution. Nature 404:388–390
Fridolfsson AK, Ellegren H (2000) Molecular evolution of the avian CHD1 genes on the Z and W sex chromosomes. Genetics 155:1903–1912
Gerrad DT, Filatov DA (2005) Positive and negative selection on mammalian Y chromosomes. Mol Biol Evol 22:1423–1432
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736
Handley LJ, Ceplitis H, Ellegren H (2004) Evolutionary strata on the chicken Z chromosome: implications for sex chromosome evolution. Genetics 167:367–376
Hellborg L, Ellegren H (2004) Low levels of nucleotide diversity in mammalian Y chromosomes. Mol Biol Evol 21:158–163
Hori T, Asakawa S, Itoh Y, Shimizu N, Mizuno S (2000) Wpkci, encoding an altered form of PKCI, is conserved widely on the avian W chromosome and expressed in early female embryos: implication of its role in female sex determination. Mol Biol Cell 11:3645–3660
Hurst LD, Ellegren H (1998) Sex biases in the mutation rate. Trends Genet 14:446–452
Itoh Y, Hori T, Saitoh H, Mizuno S (2001) Chicken spindlin genes on W and Z chromosomes: transcriptional expression of both genes and dynamic behavior of spindlin in interphase and mitotic cells. Chromosome Res 9:283–299
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammlian protein metabolism. Academic Press, New York, pp 21–132
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163
Lahn BT, Page DC (1999) Four evolutionary strata on the human X chromosome. Science 286:964–967
Lahn BT, Pearson NM, Jegalian K (2001) The human Y chromosome, in the light of evolution. Nat Rev Genet 2:207–216
Li W, Yi HS, Makova K (2002) Male-driven evolution. Curr Opin Genet Dev 12:650–656
McDonald JH, Kreitman M (1991) Adaptive evolution in gene in the ADH locus in Drosophila. Nature 351:652–654
Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T (1987) Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol 52:863–867
Montell H, Fridolfsson AK, Ellegren H (2001) Contrasting levels of nucleotide diversity on the avian Z and W sex chromosomes. Mol Biol Evol 18:2010–2016
Muller HJ (1918) Genetic variability, twin hybrids and constant hybrids in a case of balanced lethal factors. Genetics:422–499
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nicolas M, Marais G, Hykelova V, Janousek B, Laporte V, Vyskot B, Mouchiroud D, Negrutiu I, Charlesworth D, Moneger F (2005) A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol 3(1):e4
O’Neill M, Binder M, Smith C, Andrews J, Reed K, Smith M, Millar C, Lambert D, Sinclair A (2000) ASW: a gene with conserved avian W-linkage and female specific expression in chick embryonic gonad. Dev Genes Evol 210:243–249
Rice WR (1987) Genetic hitchhiking and the evolution of reduced genetic activity of the Y sex chromosome. Genetics 116:161–167
Rice WR (1996) Evolution of the Y sex chromosomes in animals. BioScience 46:331–343
Sachidanandam R, Weissman D, Schmidt SC, et al. (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, et al. (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837
Tucker P K, Adkins RM, Rest JS (2003) Differential rates of evolution for the ZFY-related zinc finger genes, Zfy, Zfx, and Zfa in the mouse genus Mus. Mol Biol Evol 20:999–1005
van Tuinen M, Dyke GJ (2004) Calibration of galliform molecular clocks using multiple fossils and genetic partitions. Mol Phyl Evol 30:74–86
van Tuinen M, Hedges SB (2001) Calibration of avian molecular clocks. Mol Biol Evol 17:451–457
Wyckoff GJ, Li J, Wu CI (2002) Molecular evolution of functional genes on the mammalian Y chromosome. Mol Biol Evol 19:1633–1636
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15:568–573
Yi S, Charlesworth B (2000) Contrasting patterns of molecular evolution of the genes on the new and old sex chromosomes of Drosophila miranda. Mol. Biol. Evol. 17(5): 703–717
Acknowledgments
We thank Dr. Lori-Jayne Lawson Handley for the turkey UBAP2W and chicken UBAP2Z and UBAP2W sequences, and two anonymous reviewers and Deborah Charlesworth for useful comments. Financial support was obtained from the Swedish Research Council. H.E. is a Royal Academy of Sciences Research Fellow supported by a grant from the Knut and Alice Wallenberg Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Deborah Charlesworth]
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Berlin, S., Ellegren, H. Fast Accumulation of Nonsynonymous Mutations on the Female-Specific W Chromosome in Birds. J Mol Evol 62, 66–72 (2006). https://doi.org/10.1007/s00239-005-0067-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0067-6