
Abstract
Adenosine deaminase (ADA) is a well-characterized enzyme involved in the depletion of adenosine levels. A group of proteins with similarity to ADA, the adenosine deaminase-related growth factors (ADGF; known as CECR1 in vertebrates), has been described recently in various organisms. We have determined the phylogenetic relationships of various gene products with significant amino acid similarity to ADA using parsimony and Bayesian methods, and discovered a novel paralogue, termed ADA-like (ADAL). The ADGF proteins share a novel amino acid motif, “MPKG,” within which the proline and lysine residues are also conserved in the ADAL and ADA subfamilies. The significance of this new domain is unknown, but it is located just upstream of two ADA catalytic residues, of which all eight are conserved among the ADGF and ADAL proteins. This conservation suggests that ADGF and ADAL may share the same catalytic function as ADA, which has been proven for some ADGF members. These analyses also revealed that some genes previously thought to be classic ADAs are instead ADAL or ADGFs. We here define the ADGF, ADAL, ADA, adenine deaminase (ADE), and AMP deaminase (AMPD) groups as subfamilies of the adenyl-deaminase family. The availability of genomic data for the members of this family allowed us to reconstruct the intron evolution within the phylogeny and strengthen the introns-late hypothesis of the synthetic introns theory. This study shows that ADA activity is clearly more complex than once thought, perhaps involving a delicately balanced pattern of temporal and spatial expression of a number of paralogous proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adenosine deaminase (ADA; OMIM 102700) catalyzes the deamination of adenosine and 2-deoxyadenosine to inosine and 2-deoxyinosine, respectively. Human ADA activity has been observed in all human tissues, due to at least three isoforms: ADA1, ADA1+CP, and ADA2 (reviewed by Hirschhorn and Ratech 1980). The ADA1 gene is located on chromosome 20q12-13 and encodes a 363-amino-acid protein of approximate molecular weight 41 kDa, and its deficiency results in one type of severe combined immune deficiency (ADA-SCID) (reviewed by Hershfield 2003). ADA1+CP is a 280-kDa protein complex composed of two ADA1 enzymes (termed “ecto-ADA” in this location) bound by the membrane glycoprotein CD26 to degrade extracellular adenosine (Franco et al. 1998). Extracellular adenosine acts through cell-specific adenosine receptors to produce different physiological effects, and therefore its concentration must be tightly regulated (Franco et al. 1997), although it is not yet known how ecto-ADA localizes extracellularly (Cordero et al. 2001). Evidence suggests that ecto-ADA can be anchored to the cell membrane by the A1R adenosine receptor as well, in order to downregulate the signal produced by adenosine (Franco et al. 1998). Interestingly, in all types of rodent cells studied, CD26 does not interact with ecto-ADA, and significant amounts of A1Rs are not expressed in hamster cells, suggesting that different mechanisms exist in rodent cells (Franco et al. 1998).
The third isoform, ADA2, is a 114-kDa molecular weight dimer that has different kinetic properties and tissue distributions compared with the other two forms, suggesting that it is coded by a separate gene of unknown structure and chromosomal location (Ungerer et al. 1992). ADA2 is found in ADA-SCID patients, proving that it results from a separate gene (reviewed by Hirschhorn and Ratech 1980). ADA2 may be produced by monocytes, since it makes up 18% of the total ADA activity in these cells (Ungerer et al. 1992), and ADA2 represents the major ADA activity in human serum (Hirschhorn and Ratech 1980), which suggests that it is secreted. This form of ADA has been found in various tissues including liver and spleen, although its proportion of the total activity in these tissues is lower (12% and 2%, respectively) compared with the other forms (24% and 86% for ADA1, 59% and 10% for ADA1+CP) (Van der Weyden and Kelley 1976). Since the specific activity of the three forms of ADA is different depending on the tissue examined (Van der Weyden and Kelley 1976), this suggests that they may have different expression patterns and therefore might be compensating for each other.
The three-dimensional structure of mouse ADA has been resolved and displays an α/β-barrel structure with a zinc atom within the active site thought to bind the activating water molecule (Wilson et al. 1991; Wang and Quiocho 1998). Several important residues have been identified as contributing to the ADA activity in the mouse protein studied. His15, His17, His214, and Asp295 are thought to be important for zinc binding, while His17, Gly184, Glu217, His238, and Asp296 are important for donating or accepting a hydrogen bond within the active site (Wilson et al. 1991; Chang et al. 1991; Sideraki et al. 1996; Mohamedali et al. 1996). Ser265 may form a salt link with His238 (Wilson et al. 1991). The α/β-barrel structure of ADA is also shared with adenine deaminase (ADE), which catalyzes a mechanistically similar deamination, converting adenine to hypoxanthine (Ribard et al. 2003). Another similar reaction, the formation of IMP from AMP, is performed by AMP deaminase (AMPD), and all three enzymes have been suspected to be related through evolution (Becerra and Lazcano 1998). There have been three types of AMPDs found with specificity in a particular tissue: AMPD1 in muscle, AMPD2 in liver, and AMPD3 in erythrocytes (Gross 1994). ADE and AMPD also share some of the ADA active site residues (Ribard et al. 2003; Wilson et al. 1991). Only prokaryotic and fungal ADEs have been discovered, presumably because higher organisms do not require the ADE function (Ribard et al. 2003).
A growing family of novel growth factors with sequence similarity to ADA has been identified. In invertebrates, this family has been termed ADGF (adenosine deaminase-related growth factor), while in vertebrates it is known as CECR1 (cat eye syndrome critical region protein 1). Various features of this family have been reviewed recently (Akalal et al. 2004). Its members include S. peregrina insect-derived growth factor (IDGF) (Homma et al. 1996), now renamed S. peregrina ADGF-A (Zurovec et al. 2002); A. californica mollusk-derived growth factor (MDGF) (Akalal and Nagle 2001); G. morsitans tsetse salivary growth factors (TSGF-1 and -2) (Li and Aksoy 2000); L. longipalpis salivary gland ADA (LuloADA) (Charlab et al. 2000); the six Drosophila homologues (ADGF-A, -A2, -B, -C, -D, and -E) (Matsushita et al. 2000; Maier et al. 2001; Zurovec et al. 2002); C. quinquefasciatus salivary ADA (Ribeiro et al. 2001); A. aegypti salivary ADA (Valenzuela et al. 2002); H. sapiens CECR1 (Riazi et al. 2000); S. scrofa CECR1; and D. rerio CECR1 (Maier et al. 2001). Note that several insect homologues have been incorrectly named “ADA” instead of “ADGF,” which will become apparent in the results. CECR1 is a candidate gene for cat eye syndrome, which is a rare human disorder characterized by defects of the eyes, heart, anus, kidneys, face, and mental development (Schinzel et al. 1981) caused by the duplication/triplication of a region of chromosome 22q11 (McDermid et al. 1986).
Some ADGF members, including S. peregrina ADGF-A (Homma et al. 2001), L. longipalpis ADA (Charlab et al. 2000; Charlab et al. 2001), A. californica MDGF (Akalal et al. 2003), and two Drosophila homologues (ADGF-A and -D) (Zurovec et al. 2002) have been shown to possess ADA activity. Salivary extracts from G. morsitans (Li and Aksoy 2000), C. quinquefasciatus, and A. aegypti (Ribeiro et al. 2001) have been shown to possess ADA activity, but the molecule responsible has not been directly identified. Since at least some ADGF proteins exhibit ADA activity, and the cytological location of ADA2 is yet to be found, there may be a connection between these two protein groups.
Although we and others have previously published the phylogenetic relationship of ADGF to ADA (Maier et al. 2001; Zurovec et al. 2002; Akalal et al. 2003, 2004), only a few ADA sequences were included as the outgroup in these analyses, which may have skewed the results. For example, our work indicates that “D. melanogaster ADA” does not belong to the classic ADAs. Also, with the ever-increasing amount of genomic and EST sequence data now available, there is a wealth of information from which to extract homologous sequences, such that a comprehensive phylogenetic analysis may be undertaken. In this study, we endeavored to find or predict protein sequences related to the ADGF and ADA genes from as many taxa as possible. In doing so, we discovered a novel group of closely related sequences, which we have called ADAL (ADA-like), and through phylogenetic analysis we show that this novel group is closely related to the classic ADAs. An analysis of conserved residues required for ADA activity showed that both the ADGF and ADAL subgroups have all the required residues for ADA activity.
Materials and Methods
Gene Discovery, Prediction, and Annotation
Protein sequences were obtained from the NCBI database using systematic BLAST searches (http://www.ncbi.nlm.nih.gov/BLAST ) (Altschul et al. 1990). In particular, the tBlastn algorithm was used with one of the subfamily protein sequences to search the GenBank (Benson et al. 2004) nonredundant (nr), EST, or species-specific (both finished and incomplete) genomic databases for similar gene products. As they were discovered, gene products were named according to sequence similarity to other proteins and/or numbered in the order in which they were found, and are included in Table 1. Our database of proteins was finalized in August 2004.
Some cDNA sequences were found in their entirety without any manipulation by the authors. Many others were predicted de novo from genomic DNA by comparison to the respective human protein, gene prediction using GENSCAN (bioweb.pasteur.fr/seqanal/interfaces/genscan.html), and/or manual extraction of the nucleotide sequence and assembly based on the tBlastn result (labeled A in Table 1). Some putative proteins were already predicted from genomic data by genome curators and placed in the database. A subset of these appeared to be correctly predicted (labeled C in Table 1), while another subset of predicted proteins appeared not to be entirely correct based on comparison to other subfamily members and were altered using an assembly of EST data or GENSCAN predictions and/or by subfamily comparison in order to obtain a better prediction (labeled B in Table 1). Only sequences that could be predicted in their entirety were included in the analysis.
Where possible, EST sequences were obtained in order to lend proof of expression of predicted genes, using tBlastn of the predicted protein against the species-specific EST database. Although there were no ESTs in the database, the expression of the D. rerio CECR1-2 gene was confirmed through RT-PCR, although the full sequence has not yet been obtained (Fang Yang, unpublished data).
The ExPASy Proteomics Server (http://kr.expasy.org) suite of programs (Gasteiger et al. 2003) was used for translation (Translate tool), molecular weight prediction (Compute pI/Mw), signal sequence prediction using SignalP (Bendtsen et al. 2004), and cellular localization using TargetP (Emanuelsson et al. 2000).
RT-PCR and Sequencing
Total RNA was isolated from adult Xenopus laevis spleen using the Trizol (Invitrogen) method, treated with DNaseI, and used with the ThermoScript RT-PCR System (Invitrogen), as per the manufacturer’s instructions. An aliquot was used in a PCR reaction with specific primers to close the gap in clone XL044i14 (accession no. BJ040131).
Sequencing of RT-PCR products and cDNA clones was carried out on an ABI 377 automated sequencer (Applied Biosystems Inc.) using vector or clone-specific primers along with the DYEnamic ET Terminator Cycle Sequencing kit (Amersham Biosciences). GeneTool v2.0 (Biotools Inc.) was used for sequence trace analysis.
Multiple Alignment and Phylogenetic Analysis
Protein sequences were aligned using MUSCLE ( http://www.drive5.com/muscle) (Edgar 2004). Alignments were checked by eye in MacClade 4.03 (Maddison and Maddison 1989), where manual editing and removal of regions with large gaps (stretches of sequence without counterparts in other species) was accomplished. Identical/similar amino acids within the alignment were shaded using BOXSHADE ( http://www.ch.embnet.org/software/BOX_form. html [K. Hofmann & M.D. Baron, unpublished]). The final alignment is available upon request to the authors.
Bayesian inference was performed using MrBayes v3.0 (Ronquist and Huelsenbeck 2003) with the Jones model (Jones et al. 1992) of amino acid substitution provided in the package. Prior probabilities for all trees were equal. The Markov chain Monte Carlo (MCMC) sampling was performed with one cold and three heated chains that were run for 550,000 generations (every 100th was saved) with a burn-in of 50,000 generations, resulting in 5000 tree samples.
Maximum parsimony (MP) analysis was performed using PAUP* version 4.0b8 (Swofford 2001). A simple amino acid substitution matrix was employed, which counts the minimum number of nucleotide substitutions required to convert one amino acid to another. The matrix of substitution was formulated by Warren Gallin, University of Alberta, based on the PROTPARS model described in Felsenstein’s PHYLIP manual version 3.6 (Felsenstein 2000). A heuristic search was performed to find the most parsimonious tree using the default parameters, except that 100 search replicates were performed, each started by random stepwise addition of taxa, before branch-swapping using tree bisection–reconnection (TBR). A bootstrap analysis of 100 replicates, each starting from 20 random stepwise additions of taxa, was performed in order to obtain support values for placement on the most parsimonious tree. Due to the large number of taxa, the bootstrap analysis took approximately 3 weeks.
Mapping of Intron Positions
For all ingroup sequences (predicted or confirmed) discovered, the cDNA sequence was compared against genomic sequence, where available, to determine the locations of exon/intron boundaries within the coding sequence. Introns located outside of the ORF were not considered. The intron positions were placed on the alignment of ingroup proteins and a matrix was built based on the presence/absence of a given intron location in each protein sequence. Introns were considered homologous only if they were identical in both location and phase. The ancestral state of each intron position was reconstructed on the rooted Bayesian topology using the “trace character” feature of MacClade (Maddison and Maddison 1989).
Results
Identification of Protein Sequences for Use in the Phylogenetic Analysis
All amino acid sequences integrated into this analysis were derived from the literature, our own work, or online databases (see Materials and Methods). We discovered two new ADGF members. A Xenopus laevis EST clone with amino acid similarity to human CECR1 was obtained from N. Ueno, National Institute for Basic Biology, Okazaki, Japan. A gap in the sequence corresponding to human CECR1 exon 3 was filled by RT-PCR (accession no. AY902778). A full-length chicken (Gallus gallus) EST clone with similarity to human CECR1 was obtained from H. Lillehoj, Animal Parasitic Diseases Laboratory, Beltsville, Maryland, USA, and fully sequenced by F. Yang (accession no. AY902779).
No mouse equivalent to human CECR1 exists (Maier et al. 2001), however, a full-length mouse EST (accession no. BC052048) was discovered with slight protein similarity
(40%) to the C-terminal region of the human CECR1 protein. This mouse protein showed slightly more similarity (41% over the entire length of the protein) to ADA and was therefore termed ADA-like (ADAL). The sequence of this full-length clone, as deposited in the database, was confirmed (R. Zurch and T. Yobb). A human ADAL homologue was discovered on chromosome 15 and its expression was confirmed by RT-PCR (M. Kardel and N. Fairbridge, unpublished results). The discovery of these two ADAL proteins spearheaded the discovery of ADAL homologues in various other organisms by the techniques described in Materials and Methods. We also scoured the databases for novel ADGF homologues. Some of the putative genes predicted from genomic DNA had at least one EST in the database to support the existence of the gene (Table 1). ADA protein sequences were also collected in silico from organisms with ADGF or ADAL representatives, in order to make the phylogenetic analysis more complete. Therefore, there are three distinct protein subfamilies with significant sequence similarity to each other: ADGF, ADAL, and ADA.
Prediction of Signal Peptides
Some members of the ADGF subfamily have been shown to be secreted. We determined the predicted cellular location of all members of the ADGF, ADAL, and ADA protein subfamilies, using signal and/or cellular localization prediction software. Most of the ADGF members were predicted to have a signal peptide (Table 1). The Drosophila species ADGF-B and -E proteins were predicted to be targeted to the mitochondria, a fact that is further strengthened by the genomic structural similarities shared between the Drosophila ADGF-B and -E genes (Maier et al. 2001). The predicted cytological location of A. gambiae ADGF-1 and -3 could not be determined, while Drosophila ADGF-A2 is suspected to be a transmembrane protein (Matsushita et al. 2000). The D. discoideum ADGF protein was predicted to contain a signal peptide, whereas the fungal ADGF proteins were not, perhaps because the fungal organisms exist as single cells. In D. discoideum, the amoeboid cells aggregate and can form a multicellular fruiting body during starvation conditions (Weijer 2004).
As expected, none of the ADA proteins were predicted to contain a signal sequence, since ADA is a cytosolic protein (Franco et al. 1998). Also, none of the ADAL proteins were predicted to contain a signal sequence, suggesting that this group of proteins may be more closely related to the ADA subfamily than to the ADGFs.
Alignment of Protein Sequences
In order to address whether the ADGF, ADAL, and ADA gene subfamilies were evolutionarily related, several phylogenetic analyses were undertaken. Since the DNA sequences showed no significant similarity between the three subfamilies, the putative protein products were compared. Also, since adenine deaminase (ADE) and AMP deaminase (AMPD) share a common reaction mechanism with ADA (Becerra and Lazcano 1998), several representative members of these two subfamilies were included in the phylogenetic analysis, to better resolve the inferred tree (see Table 1). Since two groups of adenine deaminases have evolved independently from two different ancestral proteins (Ribard et al. 2003), we only used the group of ADEs with sequence homology to the ADA subfamily for the phylogenetic analysis. E. coli ADE belongs to the group that does not share sequence similarity with ADA, and therefore does not appear in the analysis. Although there were several vertebrate AMPDs discovered in the database, only prokaryotes and fungi possess ADE (Ribard et al. 2003). In this paper, we use the definition of subfamily and family as outlined previously (Riveros-Rosas et al. 2003) and therefore describe the ADGF, ADAL, ADA, ADE, and AMPD subfamilies as belonging to the adenyl-deaminase family.
An initial alignment was constructed with all 95 protein sequences from the five subfamilies listed in Table 1. There were eight highly conserved regions found throughout the alignment of the five subfamilies, mainly focused around the catalytic residues required for ADA activity. A region was included if it was composed of at least three contiguous conserved residues, with at least two of the residues showing conservation in most members of at least three subfamilies. In order to focus on these eight important regions, the conserved amino acid residues were shaded by BOXSHADE and presented in Fig. 1. Since functional importance is highly correlated with evolutionary conservation (Gu 2001), the residues that are conserved among the five different subfamilies might indicate functional importance for the deamination process. The phylogenetic analysis (presented below) showed the AMPD subfamily as a natural outgroup of the four remaining groups, and the following observations are discussed in light of this fact. All residue numbers discussed hereafter within the alignment refer to amino acid positions within the mouse ADA protein sequence (Wilson et al. 1991) unless otherwise stated.

Protein alignment and conserved domains among the five protein subfamilies. The eight highly conserved domains from the alignment of all 95 protein sequences are displayed. Black background indicates amino acid sequence identity, while gray regions indicate conservative substitutions. Species abbreviations are as noted in Table 1. Horizontal lines delineate boundaries between individual protein subgroups. The amino acids important for ADA activity (*) are numbered underneath according to mouse ADA (His15, His17, Gly184, His214, Glu217, His238, Asp295, and Asp296). The marked ($) Arg101, Glu260, and Ser265 residues are discussed in the text.
Within the first domain, the ADGF subfamily shares a motif consisting of methionine (or iso/leucine), proline, lysine, and glycine (MPKG), the beginning of which corresponds to position 9 in the mouse ADA protein. The ADAL and ADE proteins share a conserved leucine or methionine in the first position, and both the PK residues, but not the glycine in the fourth position. The ADA proteins only conserve the PK residues of this motif, except E. coli ADA. The conservation of the proline and lysine residues throughout the ingroup suggests that these residues are important for the function of these proteins, but their role in ADA activity has not been demonstrated (Wilson et al. 1991; Sideraki et al. 1996; Mohamedali et al. 1996). The fact that the glycine is common only to the ADGF proteins suggests that it may perform a critical function only in this subfamily. All together, the AMPD subfamily seems to have retained some remnants of the full MPKG motif, but this domain was clearly not conserved over time in this group. The two ADA active site residues, His15 and His17, located at the end of conserved domain 1 are almost completely conserved among all ingroup proteins, but not within the AMPD outgroup. These two histidines are thought to be important for zinc binding (Wilson et al. 1991; Mohamedali et al. 1996). The leucine residue just previous to these important histidines is also mostly conserved throughout the ingroup, with the exception of the fungal ADEs, suggesting it may be important as well. Again, remnants of these three residues are observed in the outgroup, suggesting they were not important in the function of this subfamily. Asp19 is not conserved in proteins outside the ADA subfamily, although it was suggested to be important in the activity of ADA (Wilson et al. 1991), and was therefore not included within domain 1.
Domain 2 within the ingroup consisted of a total of nine residues: five conserved residues alternating with four less conserved sites. The last two alternating residues, glutamate (Glu; E) and arginine (Arg; R), are conserved within the entire alignment, except where Arg was changed to phenylalanine (Phe; F) for the ADEs. This last Arg residue of this domain corresponds to Arg101 in the mouse ADA protein, which is thought to form a salt bridge with Glu260, an interaction that may be important for stability (Wilson et al. 1991). The third domain is generally conserved within the ADGF, ADAL, and ADA groups, and the Gly184 residue that is important for ADA activity (Wilson et al. 1991; Sideraki et al. 1996; Mohamedali et al. 1996) is completely conserved throughout all three subgroups, except the three Drosophila ADGF-A2 proteins. Instead of the glycine residue in this position, the ADE proteins have a serine (Ser; S) and while some AMPDs have a serine in this position, others do not. Since this residue is not conserved in the AMPD and ADE groups, it might be important only for the adenosine substrate, although this has not been confirmed. The fourth and fifth domains are generally conserved throughout the entire alignment, although conservation in the AMPDs (for both domains) and the ADALs (for domain 4) is less strict. The three important residues within these two domains, His214, Glu217, and His238, are highly conserved with only a few exceptions in some of the insect ADGFs. But since the insects seem to have an overabundance of ADGF proteins (the Drosophila species harbor six ADGF proteins, and A. gambiae has at least four ADGFs), this suggests that perhaps not all of these proteins are functional or that some paralogues might have a different activity. Indeed D. melanogaster ADGF-E has previously been described as lacking ADA activity (Zurovec et al. 2002). Domain 6 is composed of a number of residues that are highly conserved throughout the entire alignment. Particularly, Glu260 and Ser265 have been suggested to form salt bridges with Arg101 and His238, respectively (Wilson et al. 1991). Glu260 is conserved in all ADGF, ADAL, and ADA proteins. Ser265 is conserved in every sequence except in two fungal ADALs, but these two proteins share a serine residue one position upstream, which may perform the same function. The seventh domain consists of only two important ADA active site residues, Asp295 and Asp296, and a proline (P) generally conserved throughout the alignment except for the ADALs. The final domain begins at mouse ADA residue 325, and although it is conserved more within the AMPD and ADGF subfamilies, its relevance is not known.
Initial Phylogenetic Inference
An initial Bayesian analysis was performed using the alignment containing all five protein subfamilies, and the consensus tree of the 5000 trees sampled was large and complex, due to the number of taxa involved. A simplified version of the tree was constructed by removing individual taxa from the tree to leave the overall relationship between the five protein subgroups. As shown in Fig. 2A, the ADAL proteins clearly form a cluster with the ADA and ADE subgroups, although much phylogenetic change has occurred between the latter groups, as represented by the long branch connecting the ADEs to their common ancestor. The phylogenetic relationship of ADA and ADE has already been established in the literature (Ribard et al. 2003), but the ADAL subfamily is a novel addition. The ADGF subfamily is distantly related to the previously mentioned groups, but the AMPD members are most distant. This indicated that the AMPD subfamily seemed to be a natural outgroup to all the other proteins and allowed the tree to be rooted from the node that the AMPDs originated from (see below). Note also that the tree topology correlates with the size of the proteins. The AMPDs have an average amino acid length of 746 ± 64 (SD), the ADGF subfamily had an average length of 531 ± 34, while the ADAL, ADE, and ADA groups had lengths of 351 ± 9, 351 ± 10, and 359 ± 15, respectively. Based on the assumption that the AMPD subfamily was the outgroup, and due to the added complexity in the alignment when these larger proteins were included, the AMPD subfamily was excluded from further in-depth analyses of the ingroup.

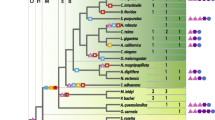
Phylogenetic analysis of the protein subfamilies using MrBayes. The scale bars in these figures represent 0.1 substitutions per site. A Initial Bayesian analysis of the five groups of protein sequences. This is a simplified representation of the tree inferred using all of the ADGF, ADAL, ADA, ADE, and AMPD proteins, showing the AMPD group as a natural outgroup. All taxa from each group were clipped from the tree and replaced by a triangle, whose width is proportional to the number of taxa in that group. B Phylogenetic analysis of the ingroup. This unrooted tree was inferred using Bayesian analysis on the alignment of the ADGF, ADAL, ADA, and ADE gene products. Species abbreviations are as noted in Table 1. The arrow indicates the approximate location of the root, if the AMPD outgroup had been included (see A for confirmation). Horizontal lengths of branches are proportional to the estimated numbers of amino acid substitutions. The values on top of the internal branches indicate the posterior probability that the clade is correct under the model, summed over 5000 tree samples, and are depicted as percentages. One node (indicated as a polytomy) had a posterior probability less than 50% in the Bayesian analysis. Numbers indicated below the branches in boldface italics are MP bootstrap proportions shown for comparison (see Supplementary Fig. S1). Bootstrap values that were identical to the Bayesian probabilities are not included here.
Focused Analysis of the Ingroup
A second alignment of the 80 protein sequences belonging to the ingroup (ADGF, ADAL, ADA, and ADE) was used for further phylogenetic analyses. Bayesian analysis was run five separate times (550,000 generations each) and the resulting tree topology was identical each time. Between the five MrBayes runs, the posterior probabilities (support values) at each node varied between runs by up to 5% in most cases, which is expected with this sampling methodology. The first analysis was chosen as a representative of the five runs, and its associated branch lengths and posterior probabilities are presented in Fig. 2B. Two major clades were evident in this tree: (1) ADAL, ADE, and ADA and (2) ADGF. This major split between the two groups was well supported, as indicated by the posterior probability value of 1.00 (represented as a percentage in the figure). This major split was also observed in the initial analysis that included the AMPD protein sequences, and the approximate placement of the “ROOT” between these two groups in Fig. 2B represents this outgroup. Overall, the entire topology was well supported, especially for many of the deep divergences, with only a few weak posterior probabilities for some of the internal nodes.
Within the first major clade, support was high for the split between the ADAL and the ADE/ADA groups (100 and 99%, respectively), but support for some internal nodes within each group was problematic. Bayesian analysis tends to give high posterior probabilities for internal nodes, such that a value less than 70% might be considered to be low (Huelsenbeck et al. 2001). Support values for the nodes leading to C. elegans ADAL, U. maydis ADAL, and the two pufferfish ADALs (T. rubripes and T. nigroviridis) were <50, 63, and 64%, respectively, suggesting that although these proteins clearly belong to the ADAL subfamily, there was a lack of confidence for their placement within the subfamily. Also, the placement of these taxa disagrees with the accepted phylogenetic relationship of organisms as known to date ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db= taxonomy), which shows that U. maydis ADAL should form a clade with the two other fungal ADALs, and the pufferfish ADALs should appear basal to X. laevis ADAL. Support for the inclusion of E. coli ADA in a monophyletic group with the other ADAs was only 51%, as this protein sequence was often grouped with the ADE subfamily. Support was also less than optimal in clades containing T. nigroviridis ADA and G. gallus ADA, since the posterior probabilities for these clades were in the low 70s.
Within the second major clade, the ADGF proteins fall into several distinct subgroups that were very well supported: fungi, vertebrates, and insects. A. californica MDGF appears basal to the insects, while D. discoideum ADGF is basal to both the vertebrates and the insects. The general scheme of this major clade agrees with the accepted organismal phylogeny, except that D. discoideum should appear basal to the entire group, yet this bipartition was found in only 6% of the sampled trees. Note that there are several duplication events within this major clade that occurred after these groups branched from the common phylogenetic tree. All the fungi seem to have two ADGF homologues, except U. maydis, which either has lost one copy or has yet to be sampled. The fish CECR1 genes have also undergone a further subdivision, which may be a result of the tetraploidation of ray-finned fish (reviewed by Taylor et al. 2003). Instead of forming a clade with the other fish CECR1-1 proteins, D. rerio CECR1-1 appeared basal to the entire group, indicating that perhaps this gene has retained more of the ancestral features than the other genes. Within the insect subgroup, the six Drosophila ADGF gene products act as a backbone onto which the other insects with less fully sequenced genomes may be placed. For example, only one ADGF family member has been discovered in the flesh fly, Sarcophaga peregrina, and this protein groups with the Drosophila ADGF-A members. Sequencing of the entire S. peregrina genome may reveal five other gene products similar to the other ADGF members present in the Drosophila species. There was low support (56%) for the clade containing A. aegypti ADA and A. gambiae ADGF-1 due to the almost equally probable topology where A. gambiae ADGF-1 and -3 form a monophyletic group (44%). This might indicate that a duplication occurred in the A. gambiae lineage after diverging from the other organisms, but this problem will be better resolved once the A. aegypti genome is completely sequenced and more ADGF sequences are discovered.
Overall, it seems that the protein names given to sequences found in the tBlastn searches were correct. For example, all the proteins that were named ADAL are most closely related to each other, without any appearances in other subfamilies. Some protein sequences that were labeled previous to this study and/or published by other groups may in fact be mislabeled. D. melanogaster ADA, L. longipalpis ADA, C. quinquefasciatus ADA, and A. aegypti ADA do not group with the classic ADA proteins. Instead, D. melanogaster ADA is a member of the ADAL subfamily, while L. longipalpis ADA, C. quinquefasciatus ADA, and A. aegypti ADA belong to the insect ADGFs.
Parsimony Analysis
In order to check the accuracy of the ingroup topology produced by Bayesian analysis, a maximum parsimony (MP) analysis was performed on the same ingroup alignment. A heuristic search recovered only one most parsimonious tree, however, bootstrap support on this tree was not very robust, especially for many internal nodes, such that 14 of the 77 bootstrap values were less than or equal to 50% as placed on the most parsimonious tree (see Supplementary Fig. S1). Whereas a value of 70% might indicate strong support for a group, since bootstrap proportions are conservative measures of support (Holder and Lewis 2003), the general lack of support for the most parsimonious reconstruction indicated that perhaps the use of MP for this data set was not optimal for resolution of internal nodes.
Importantly, however, the subgroups and major clades found in the Bayesian tree were also retained in the MP tree, and the bootstrap support for these clades was high, as shown in Fig. 2B. Some internal nodes of the MP tree that were not well supported by bootstrapping were also not well supported with Bayesian posterior probabilities, such as the placement of U. maydis and C. elegans ADAL within the ADAL subgroup. Also, E. coli ADA was placed equally within either the ADE or the ADA subgroup for both analyses (see Fig. 2B). Within the ADGF clade, however, there was a major mismatch in the MP tree compared to the Bayesian result. In the MP tree, the A. californica MDGF and vertebrate ADGF proteins form a sister group to the insect ADGF-C and -D proteins, with the insect ADGF-A, -B, and -E as the sister to those groups. Also, the ADGF-A2 proteins, instead of being placed as a sister group to the ADGF-A, -B, and -E clade as in the Bayesian analysis, were placed as a sister group to all other vertebrate and insect ADGFs. In effect, the vertebrate proteins were nested within the insect homologues in the MP tree, which produces a major conflict with the Bayesian and established organismal trees. This topology, however, was associated with very low bootstrap values on the most parsimonious MP tree (see Supplementary Fig. S1).
The nesting of the CECR1 vertebrates within the ADGF insects was obtained previously by our group using a maximum likelihood approach (Maier et al. 2001) but was not observed in any of the five final MrBayes runs. In order to determine if this topology in fact was more probable but had just been overlooked (not sampled) by the Bayesian analysis, we ran MrBayes using the parsimony result as a user- defined starting tree. If the MP tree was more highly probable than that previously obtained by MrBayes, then this topology would be expected to persist throughout the run. Instead, the original MrBayes result was again obtained. Also, since the support value for the insect ADGF clade in the Bayesian tree shown in Fig. 2B was 100%, this indicated that there were no instances in which the MP topology that included the ADGF vertebrates was observed, suggesting that the MP topology is not highly probable.
Intron Evolution in This Large Protein Family
Due to the wealth of genomic sequence available, we used the results of our phylogenetic analysis to determine the evolution of intron positions among the four ingroup subfamilies. After mapping all intron positions onto the alignment of ingroup proteins, there were a total of 52 distinct intron positions observed in at least one protein sequence. Each intron position was coded into the matrix presented in Fig. 3, which was then used to reconstruct the most parsimonious intron gain/loss pattern on the inferred Bayesian topology (see Fig. 3). Only the accelerated transformation reconstruction is shown, in order to more directly test the earliest appearance of each intron. For example, intron location 18 is shown in the figure to be gained in the ancestor of some of the fungal ADGF-1 proteins and then lost in M. grisea ADGF-1. If the changes were delayed instead of accelerated, there would be two instances of intron 18 gain, in both G. zeae ADGF-1 and N. crassa ADGF-1. Both situations require the same number of steps and are therefore equally parsimonious, but only the accelerated reconstruction is presented. Other introns with an equal number of delayed reconstruction steps include positions 8, 15, 37, and 46.

Reconstruction of introns gained or lost within the ingroup phylogeny. Left A cladogram of the Bayesian topology depicted in Fig. 2B with intron gain/loss mapped onto it. Species abbreviations are as noted in Table 1. Some taxa (*) lack genomic data and therefore the intron status along these terminal branches is unknown. Numbers refer to intron positions within the alignment that have been gained (+) or lost (–), according to the most parsimonious reconstruction (least number of steps). Intron positions in boldface involve more than one step and are therefore found more than once in the figure. Right A table of occurrences of the 52 intron positions within the ingroup alignment, from which the reconstruction on the left was derived. The presence (+), absence (–), or unknown status (?) of each intron position is indicated beside each taxa.
In general, while none of the bacterial or ADE genes had introns, the fungal and insect genes had between 0 and 5 introns, and the vertebrates had 8 to 11 introns, suggesting that organisms that have diverged more recently tend to have more introns. Throughout the entire ingroup, 14 of the 52 intron positions were found in only one taxa. Many of the remaining shared intron positions are found on the branches leading to the vertebrate groups, since five intron positions are found only within the vertebrate ADALs, eight positions within the vertebrate ADAs, and five positions in the vertebrate ADGF homologues (CECR1s). This leaves only 20 intron locations shared among other organisms. Surprisingly, only one intron was found in the exact same place between two different subgroups (position 44; N. crassa ADAL and M. grisea ADGF-1), but since it was present in only one member of each subgroup, it is most probably a coincidence rather than the persistence of a common intron position in an ancestor of the entire ingroup. Within the ADGF subfamily, positions 19 and 37 are faithfully conserved between the vertebrates and some of the insect subgroups, but this would only suggest that the intron was present in a common ancestor of the metazoans.
Some intron positions between different subgroups are located within just a few base pairs of each other, which might suggest that they have originated in a common ancestor of those subgroups. This suggestion relies on the theory of intron-sliding, which is the apparent shift of an entire intron by only a few base pairs between two taxa (reviewed by Stoltzfus et al. 1997). Spliceosomal introns are not self-splicing and are not known to be mobile, so the loss or gain of an intron in a specific lineage is likely to be a unique event (Venkatesh et al. 1999). Therefore, it might be more parsimonious for the intron to slip rather than be lost and then gained in a nearby location (Schmitt and Brower 2001). Introns 4–7 may have originated from a common ancestor, when intron-sliding is taken into account. These four positions are separated by only 9 bp total and are associated with Domain 1 in the ingroup alignment (see Fig. 1). Intron position 4 is located in the vertebrate ADAs just after the conserved “PK” residues, and intron position 5 lies within the G of the “MPKG” motif conserved throughout the vertebrate ADGF homologues. Due to the gap introduced in the ADAs in lieu of this G residue, intron 4 position is actually embedded within the location of intron 5. Intron position 6 is located one codon further, between the vertebrate and A. gambiae ADAL “VE” residues, while position 7 is again one codon further, precisely after the “VE” residues, in the fungal ADALs. Even when intron-sliding is taken into account, however, if this intron position did indeed originate in a common ancestor of the four subgroups, the position would need to be lost at least 10 different times to account for its absence in all the other ingroup proteins. In this case, it might be more parsimonious to gain the location four times, suggesting that even if intron-sliding is considered, positions 4–7 probably arose independently.
There are three more cases where intron positions might be conserved between different subgroups when intron-sliding is considered. Intron positions 20–22 are separated by only 5 bp in most cases and involve both the ADA and the ADAL vertebrates with the intervening position 21 located in C. elegans ADA, but the position would need to be lost at least six times on other branches. Interestingly, intron position 36 found in the vertebrate ADALs is located only 4 bp upstream of position 37, which was described above to be shared between the vertebrate ADGFs and the insect ADGF-C and -D clade, but again, if this intron were present in a common ancestor of the ingroup, it would need to be lost eight separate times. Finally, position 41 in the ADAL vertebrates is located 2 bp upstream of position 42 found in the vertebrate ADAs but would need to be lost seven times, which again does not represent the most parsimonious reconstruction.
Within one subgroup, two groups of intron positions may have arisen through intron-sliding. Intron position 29, found in the insect ADGF-C and -D clade, is located just 1 bp before position 30, which is found in the insect ADGF-A2, -A, -B, and -E clade. But in this case, two separate instances of intron gain may be more parsimonious than gain of this intron, sliding to position 30, and loss in the clade containing A. gambiae ADGF-1 and -3, which represents three separate steps. The only case in which intron-sliding might be more parsimonious occurs within the vertebrate ADAs, where all eight of the intron positions in this group are precisely conserved, except for an instance of intron-sliding in D. rerio ADA (intron 8 slid 3 bases to position 9). This event represents only two steps (gain plus slide) versus three steps (gain of position 8 in all vertebrates, followed by loss of 8 and gain of 9 only in D. rerio), suggesting that intron-sliding is a likely explanation only for this one case. Although it is difficult to decipher how many minor shifts in intron position could be explained by intron-sliding, it seems clear that for this data set, use of this theory is not helpful in suggesting that any of the observed intron positions were present in a common ancestor.
Discussion
This paper highlights the discovery of proteins comprising a novel subfamily, ADAL, with membership in the family of adenyl-deaminases. We have also added several new members to the ADGF subfamily, although since most of these new additions are merely predictions, their existence and actual sequence still need to be confirmed. The phylogenetic analysis of this protein family showed that the ADGF and ADAL subgroups are clearly related to the classic ADA subfamily. The ADAL proteins are more closely related to the ADA and ADE subgroups, in both sequence similarity and number of residues, compared to the ADGF group. Also, many of the ADGF members have a predicted signal peptide, whereas none of the ADA or ADAL proteins do, which further confirms their similarity to each other. This raises the issue of redundancy between ADAL and ADA and, perhaps, ADGF. Why have three separate groups of proteins evolved to carry out the same apparently simple function? Although no ADAL subfamily members have been shown to have ADA activity, if it is proven that they do, there would be three subfamilies, ADAL, ADGF, and the classic ADAs, which harbor ADA activity. Perhaps ADA function has been compartmentalized, both spatially and temporally, for various tissues. Altogether, it seems that ADA activity is a much more complicated story than previously thought.
Conservation of ADA Active Site Residues
The crystal structure of mouse ADA has identified amino acid residues with a specific role in the function of the protein (Wilson et al. 1991). Comparison of the protein sequence of ADA to ADE has outlined differences that may be attributed to the deamination of adenosine versus adenine (Ribard et al. 2003). Asp19, Ser103, Ala183, and Gly184 are characteristic of ADAs, while in ADEs they are replaced by Glu, Asp, Asp/Ser, and Ser, respectively (Ribard et al. 2003). Asp19 and Ser103 are thought to bind the ribose in different places, while Ala183 and Gly184 are described to be involved in the active site. Besides Gly184, the ADEs have retained all other ADA active site residues (Ribard et al. 2003). From our work, it is apparent that Asp19 (data not shown; the residue lies outside of conserved domain 1 shown in Fig. 1) and Ala183 are not conserved within the ADGF or ADAL subfamilies, and while most of the ADALs have a synonymous Thr residue in place of Ser103 (data not shown), the ADGFs do not. This suggests that ADGF and ADAL might react with slightly different substrates, perhaps an adenosine analogue. On the other hand, we have shown that, except for some of the insects, all of the ADGF and ADAL members have retained all eight residues required for ADA activity, including the Gly184 mentioned above. The three residues involved in salt-bridge formation, Arg101, Glu260, and Ser265, are also conserved in the ADGF and ADAL subfamilies but not in ADEs.
Some ADGF members, including S. peregrina ADGF-A (Homma et al. 2001), L. longipalpis ADGF (Charlab et al. 2000, 2001), A. californica MDGF (Akalal et al. 2003), and two Drosophila homologues (ADGF-A and -D) (Zurovec et al. 2002), have been shown to possess ADA activity. Salivary extracts from G. morsitans (Li and Aksoy 2000), C. quinquefasciatus, and A. aegypti (Ribeiro et al. 2001) have been shown to possess ADA activity. Both the L. longipalpis ADA and the A. californica MDGF proteins have been modeled based on the structure of mouse ADA, which showed that all the active site residues in the two ADGF subfamily proteins were conserved in the correct structural locations (Charlab et al. 2001; Akalal et al. 2004). Therefore, this might indicate that, like all ADAs and some insect ADGFs, all the members of the ADGF and ADAL subfamilies may also harbor ADA activity. Also, the novel MPKG motif is conserved in almost all ADGFs, with the PK being conserved in both the ADA and the ADAL subfamilies, suggesting that this region of the protein may be important in the overall function, although the significance of this domain is unknown presently. Once the function of the vertebrate ADGFs and ADALs has been determined, it will be possible to compare the conserved residues within this entire family to assign specific functional roles for each residue.
Patterns Revealed in the Phylogenetic Analysis
Both the Bayesian and the MP trees showed the ADGF, ADAL, ADA, and ADE subgroups as separate, well-defined splits, which were very well supported by both analyses. The major mismatch between the two methods involving the grouping of the vertebrate ADGFs within the insects in the MP tree was tested by using that topology as a starting tree for a MrBayes analysis. Considering the fact that the MP tree was not maintained in the MrBayes run, and the lack of internal support within the MP topology, as well as the better fit of the Bayesian tree with the established organismal phylogeny, we preferred the Bayesian result to that of MP for this data set.
In general, the phylogenetic analysis revealed that some genes previously thought to be classic ADAs are more correctly placed elsewhere. D. melanogaster ADA (as named in FlyBase) is a member of the ADALs, while L. longipalpis ADA (LuloADA), C. quinquefasciatus ADA, and A. aegypti ADA belong with the ADGF subfamily. It therefore seems that these insect proteins have been incorrectly labeled. Just as it was previously suggested to rename S. peregrina IDGF to S. peregrina ADGF-A (Zurovec et al. 2002), we suggest the renaming of D. melanogaster ADA to D. melanogaster ADAL, L. longipalpis ADA to L. longipalpis ADGF, C. quinquefasciatus ADA to C. quinquefasciatus ADGF, and A. aegypti ADA to A. aegypti ADGF, in order to better reflect their position within the adenyl-deaminase family. It seems, therefore, that none of these insects has a homologue of the classic ADAs. For organisms with incomplete genomes, perhaps the classic ADA homologue has just not been found. D. melanogaster and A. gambiae, however, have complete or nearly finished genomic sequence, and a homologue to the classic ADAs has not been found in these two organisms, suggesting that is was lost in these insects. There were no classic ADA homologues found in the fungi either, whereas ADAL was found in insects, vertebrates, and most fungi but not in prokaryotes. This suggests that certain protein subfamilies may be specialized for certain organisms or that perhaps the three subfamilies (ADGF, ADAL, and ADA) are partially redundant. Also, because there were no prokaryotic orthologues of ADGF or ADAL, this suggests that these proteins were gained on the lineage leading to extant eukaryotes.
Many organisms have multiple ADGF paralogues, including the fish, fungi, and insects, which may indicate the importance of the ADGF protein for development. It is likely that some of the paralogues have acquired a specialized expression pattern. For example, D. melanogaster ADGF-B and -A2 are both male specific, while ADGF-A and -D are more universally expressed (Matsushita et al. 2000; Maier et al. 2001). The various paralogues may also have been adapted for a broader range of functions. There are six Drosophila ADGFs, and some D. melanogaster proteins have been shown to have ADA activity (ADGF-A and -D), while others, such as ADGF-E, may be nonfunctional (Zurovec et al. 2002). Since ADGF expression has been observed mainly in the salivary glands of biting insects, and has been suggested to aid insects in providing pain relief at the site of biting (Charlab et al. 2001; Li and Aksoy 2000; Ribeiro et al. 2001), perhaps ADGF has been adapted for a specialized physiological purpose in insects and other lower organisms, compared to mammals. The Drosophila species ADGF-E protein does not share four of the eight conserved ADA residues, which may explain its lack of ADA activity. The replacements for these four residues, however, are faithfully conserved between the three Drosophila ADGF-E sequences (D. yakuba ADGF-E is not shown), suggesting that this paralogue has taken on a new role. The lack of conservation of the ADA active site residues was also observed for some of the other insect ADGFs. Some of the Drosophila proteins, including ADGF-E, are predicted to be localized to the mitochondria, which again suggests the evolution of a new function. Some nonfunctional paralogues may also represent expressed pseudogenes.
The phylogenetic analysis showed that the three Drosophila species acted as a backbone onto which the other insect genes from incomplete genomes may be placed. Since there are already six paralogues within Drosophila, it seems odd that both A. gambiae and G. morsitans apparently have two genes that are more similar to each other than to the Drosophila orthologues (as shown in Fig. 2B). This suggests that, in addition to the six possible paralogues similar to Drosophila, these two organisms may have separately undergone an additional duplication event to produce a seventh paralogue. Conversely, these results might be explained by gene conversion (reviewed by Papadakis and Patrinos 1999), such that the two genes appear to be more similar to each other than to one of the other Drosophila homologues. Finally, the duplications observed in the Drosophila species might not have occurred before the divergence of all the insect species, and the one to three genes found in each of the other insect species may have been scattered on the Drosophila backbone simply according to the amount of sequence similarity in these genes. The availability of finished genome sequence for the other insect species may help to clarify this issue.
There are several family members that seem to be missing from completely sequenced genomes. There is no M. musculus, R. norvegicus, or C. elegans homologue in the ADGF subfamily, but homologues of the AMPD, ADA, and ADAL exist in these organisms. This suggests that the ADGF homologue has been lost in these organisms, and perhaps one of the other subfamily members may be compensating for the loss. Also, none of the Drosophila species or other insects seem to have an ADA homologue. Perhaps the insect lineage has lost ADA, due to the six ADGF paralogues that have presumably replaced its function. Unlike ADGF, there is only one ADA and one ADAL homologue found in all three fish species studied, indicating either that these genes were not part of the major gene duplication event (Taylor et al. 2003) or that the duplicates of these family members were lost. It is interesting that there is no S. cerevisiae homologue of any other subfamily members besides ADE, especially since S. cerevisiae ADE has been previously mistaken for a classic ADA in the literature. As mentioned previously, E. coli ADE exists but was not included in the phylogenetic analysis, and although both E. coli and S. coelicolor possess an ADA and ADE gene, no other family members were found in either bacterial species. For organisms with unfinished genomes, the lack of a certain gene product may be due to a loss of that gene in the organism or may simply mean that it has not been sequenced yet. This may be especially true for organisms with almost no genomic information, including D. discoideum, X. laevis, S. scrofa, A. californica, L. longipalpis, C. quinquefasciatus, A. aegypti, S. peregrina, and G. morsitans. Three fungal ADE members have been discovered thus far, but there have been no fungal ADAs found, and although this could be due to the unfinished state of many fungal genomes, one might expect to find an ADA homologue in at least one of the six fungal genomes searched.
Are ADGF and ADA2 the Same?
ADA2 is one of three ADA isoforms found in certain cell types, and it is especially important in human plasma, where it is responsible for the majority of ADA activity (Hirschhorn and Ratech 1980). It is likely that ADA2 is a member of the adenyl-deaminase family. A recent paper described the purification of the chicken ADA2 protein from liver extracts and showed that the 110-kDa dimer (active form) has a monomer weight of 55 kDa and is N-glycosylated (Iwaki-Egawa et al. 2004). The mature (without the predicted 23-amino-acid signal sequence) chicken CECR1 protein (ADGF member) is predicted to have a molecular weight of 55.7 kDa. Also, there are two N-glycosylation sites predicted with high confidence in the chicken CECR1 protein, at positions 172 and 295 (NetNGlyc Server; http://www.cbs.dtu.dk/services/NetNGlyc ). The first 12 N-terminal amino acids (TPLWSLMQDLMM) of chicken ADA2 were determined by Edman degradation (Iwaki-Egawa et al. 2004). The first 12 N-terminal amino acids of chicken CECR1 (TPLWEDRDSLMQ) are surprisingly similar, but not identical, to those of chicken ADA2. Note that the first and last four residues of the CECR1 N-terminus are identical to the first eight residues of the chicken ADA2 protein, suggesting that there may have been either a sequencing or a cloning error in one of the proteins. Other evidence for the comparison of chicken ADA2 to CECR1 comes from the analysis of other ADGF proteins. Human CECR1 is expressed highly in peripheral blood leukocytes (S. Maier, unpublished results), cells that might secrete CECR1 into the blood plasma. Also, S. peregrina ADGF-A was observed as a homodimer, and its ADA activity was inhibited by 2′-deoxycoformycin (DCF) (Homma et al. 1996, 2001), which also inhibits ADA2 (Niedzwicki and Abernethy 1991). Together, these observations suggest that the ADGF proteins may indeed be the identity of the elusive ADA2 protein. ADAL could not be considered for this role because its predicted molecular weight is 40.3 kDa, and it is not predicted to be secreted.
ADA-deficient mice exhibit severe pulmonary insufficiency, bone abnormalities, and kidney pathogenesis (Blackburn et al. 1998), a phenotype much worse than is found in humans. Although mouse has a copy of ADAL and ADA, there has been no ADGF homologue found in rodents, whereas humans have one member of each subfamily. Since the plasma and other tissues of mice and rats contain ADA1 but not ADA2 activity (Niedzwicki et al. 1995), this suggests that perhaps the rodents have lost ADA2. This fact represents yet another clue that the identity of ADA2 is ADGF, since they are both missing from the rodent lineage. It is currently under investigation as to where ADAL is expressed. Perhaps both the ADA and the ADAL homologues in mice are compensating for the loss of the ADGF gene product. If the human ADGF homologue (CECR1) and/or ADAL are shown to possess ADA activity, then these proteins may be contributing to the overall ADA activity of various tissues. This also could explain the relatively mild phenotype in humans deficient in ADA, which has both ADGF and ADAL proteins intact, while ADA–/– mice are more seriously affected, since only the function of the ADAL protein remains. Also, ADA and ADAL have predicted molecular weights of 40.8 and 40.3 kDa, respectively. Since their sizes are so similar, ADA activity previously attributed to ADA1, the 41-kDa form of ADA (Van der Weyden and Kelley 1976; Ungerer et al. 1992), might indeed be due to one or the other, or both. Their similarity in size may have concealed ADAL until now.
Proof for the Introns-Late Aspect of the New Synthetic Theory of Introns
If the intron position data are available, large gene families are useful for studying the relationship of evolution and the conservation of intron positions. There has been an important debate in the last few decades over whether spliceosomal introns were present in primitive coding sequences (“introns-early”) or added later (“introns-late”) in the lineage leading to eukaryotes (Gilbert et al. 1986; de Souza et al. 1998; Fedorova and Fedorov 2003). The introns-early hypothesis is supported by the observed correlation of phase 0 intron positions (inserted after the third codon position) and the separation of structural protein domains (Fedorov et al. 2001; de Souza et al. 1998). Introns-early proponents also invoke the theory of intron-sliding to suggest that intron positions located a few base pairs away in other phyla have only shifted (Stoltzfus et al. 1997). Indeed, it might be more parsimonious for the intron to slide rather than be lost and then gained in a nearby location (Schmitt and Brower 2001). Clearly though, many genes have had introns introduced in the lineage leading to eukaryotes, thus validating the introns-late side of the debate (de Souza 2003). Also, although one group concluded that intron-sliding by 1 bp might be a real evolutionary phenomenon, they suggest that it would be a relatively rare event, occurring in <5% of all introns (Rogozin et al. 2000), so the use of this theory for one or the other side of the debate may be a moot point. Recently, a new “synthetic” theory of intron evolution has been proposed, suggesting that most introns, especially those that are phase 1 or 2, are recent acquisitions of eukaryotic genes, but a subset of the present-day phase 0 introns are candidates to be ancient (de Souza 2003), thus incorporating concepts from both theories.
The ADA family has both eukaryotic and prokaryotic members and would therefore be considered ancient. The duplication and divergence of the other three groups might also be considered to be ancient, especially when considering the fungal genes found in each. But only one intron position was conserved between two of the four ingroup subfamilies when intron-sliding was not considered, and it was found in only one member of each of those subfamilies, suggesting that it might be a coincidence. If the introns-early aspect of the synthetic theory was to be accepted, it might be expected that more intron positions would be retained between the four subgroups of the ingroup. There were many instances where an ancestral intron might have existed when intron-sliding was taken into account. For each case, however, the most parsimonious reconstruction favored a few instances of intron gain rather than many more losses. A trend seemed to emerge within the organisms in this study that there are generally more introns found in higher organisms compared with the lower deuterostomes, which have an intermediate number of introns, and single-celled organisms with no introns. This suggests in general that introns were added along the eukaryotic lineage over time.
These data, although they seem to substantiate the introns-late side, have done nothing to rule out the introns-early aspect of the synthetic theory. Indeed, analysis of the intron phases within the entire ingroup showed a slight excess of phase 0 introns (46%; 145/316). Also, use of the lack of conserved introns to support the introns-late side assumes that intron loss and gain are equally likely. If intron loss was entirely easier that intron gain, the introns-early side may hold some weight with these data. Therefore, it is possible that the resolution to the debate cannot be undertaken until the relative costs of intron loss/gain are determined (Tyshenko and Walker 1997). In a study of human, coral, fly, and worm integrin-β genes, the coral gene shared 25 of 26 intron positions with at least one other species, when intron-sliding was taken into account (Schmitt and Brower 2001). Without the coral sequence, only eight splice sites were shared between two or more phyla. This suggests that without an ancestral sequence such as this coral sequence, the results might incorrectly appear to only support the introns-late aspect of the new synthetic theory. Therefore, although our results at present seem only to support the introns-late side, the addition of more data as they become available may change this view.
In conclusion, ADA activity is clearly not as straightforward as once thought. The structure and conserved residues of the ADGF and ADAL subfamilies, combined with their evolutionary relationship to classic ADAs, suggest that these three genes are all involved in ADA activity. If the expression of each is found in a variety of cellular locations, together they may control adenosine levels in a concerted fashion.
Note added in proof. The identity of human ADA2 as the CECR1 gene product has been confirmed and published by A.V. Zavialov and A. Engstrom. Their manuscript was e-published in the Biochemical Journal on June 1, 2005, and is entitled “Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity.”
References
Akalal DB, Nagle GT (2001) Mollusk-derived growth factor: cloning and developmental expression in the central nervous system and reproductive tract of Aplysia. Brain Res Mol Brain Res 91:163–168
Akalal DB, Bottenstein JE, Lee SH, Han JH, Chang DJ, Kaang BK, Nagle GT (2003) Aplysia mollusk-derived growth factor is a mitogen with adenosine deaminase activity and is expressed in the developing central nervous system. Brain Res Mol Brain Res 117:228–236
Akalal DB, Schein CH, Nagle GT (2004) Mollusk-derived growth factor and the new subfamily of adenosine deaminase–related growth factors. Curr Pharm Des 10:3893–3900
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Becerra A, Lazcano A (1998) The role of gene duplication in the evolution of purine nucleotide salvage pathways. Orig Life Evol Biosph 28:539–553
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Benson DA, Karsch–Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2004) GenBank: update. Nucleic Acids Res 32 (database issue):D23–D26
Blackburn MR, Datta SK, Kellems RE (1998) Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem 273:5093–5100
Chang ZY, Nygaard P, Chinault AC, Kellems RE (1991) Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residues: implications for catalytic function. Biochemistry 30:2273–2280
Charlab R, Rowton ED, Ribeiro JM (2000) The salivary adenosine deaminase from the sand fly Lutzomyia longipalpis. Exp Parasitol 95:45–53
Charlab R, Valenzuela JG, Andersen J, Ribeiro JM (2001) The invertebrate growth factor/CECR1 subfamily of adenosine deaminase proteins. Gene 267:13–22
Cordero OJ, Salgado FJ, Fernandez-Alonso CM, Herrera C, Lluis C, Franco R, Nogueira M (2001) Cytokines regulate membrane adenosine deaminase on human activated lymphocytes. J Leukoc Biol 70:920–930
de Souza SJ (2003) The emergence of a synthetic theory of intron evolution. Genetica 118:117–121
de Souza SJ, Long M, Klein RJ, Roy S, Lin S, Gilbert W (1998) Toward a resolution of the introns early/late debate: only phase zero introns are correlated with the structure of ancient proteins. Proc Natl Acad Sci USA 95:5094–5099
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Fedorov A, Cao X, Saxonov S, de Souza SJ, Roy SW, Gilbert W (2001) Intron distribution difference for 276 ancient and 131 modern genes suggests the existence of ancient introns. Proc Natl Acad Sci USA 98:13177–13182
Fedorova L, Fedorov A (2003) Introns in gene evolution. Genetica 118:123–131
Felsenstein J (2000) PHYLIP (Phylogeny Inference Package) version 3.6 alpha. Computer programs and documentation. Department of Genetics, University of Washington, Seattle
Franco R, Casado V, Ciruela F, Saura C, Mallol J, Canela EI, Lluis C (1997) Cell surface adenosine deaminase: much more than an ectoenzyme. Prog Neurobiol 52:283–294
Franco R, Mallol J, Casado V, Lluis C, Canela EI, Saura C, Blanco J, Ciruela F (1998) Ecto-denosine deaminase: an ecto-enzyme and a costimulatory protein acting on a variety of cell surface receptors. Drug Dev Res 45:261–268
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788
Gilbert W, Marchionni M, McKnight G (1986) On the antiquity of introns. Cell 46:151–153
Gross M (1994) Molecular biology of AMP deaminase deficiency. Pharm World Sci 16:55–61
Gu X (2001) Maximum-likelihood approach for gene family evolution under functional divergence. Mol Biol Evol 18:453–464
Hershfield MS (2003) Genotype is an important determinant of phenotype in adenosine deaminase deficiency. Curr Opin Immunol 15:571–577
Hirschhorn R, Ratech H (1980) Isozymes of adenosine deaminase. Isozymes Curr Top Biol Med Res 4:131–157
Holder M, Lewis PO (2003) Phylogeny estimation: traditional and Bayesian approaches. Nat Rev Genet 4:275–284
Homma K, Matsushita T, Natori S (1996) Purification, characterization, and cDNA cloning of a novel growth factor from the conditioned medium of NIH-Sape-4, an embryonic cell line of Sarcophaga peregrina (flesh fly). J Biol Chem 271:13770–13775
Homma KJ, Tanaka Y, Matsushita T, Yokoyama K, Matsui H, Natori S (2001) Adenosine deaminase activity of insect-derived growth factor is essential for its growth factor activity. J Biol Chem 276:43761–43766
Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP (2001) Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294:2310–2314
Iwaki-Egawa S, Namiki C, Watanabe Y (2004) Adenosine deaminase 2 from chicken liver: purification, characterization, and N-terminal amino acid sequence. Comp Biochem Physiol B Biochem Mol Biol 137:247–254
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Li S, Aksoy S (2000) A family of genes with growth factor and adenosine deaminase similarity are preferentially expressed in the salivary glands of Glossina m. morsitans. Gene 252:83–93
Maddison WP, Maddison DR (1989) Interactive analysis of phylogeny and character evolution using the computer program MacClade. Folia Primatol 53:190–202
Maier SA, Podemski L, Graham SW, McDermid HE, Locke J (2001) Characterization of the adenosine deaminase–related growth factor (ADGF) gene family in Drosophila. Gene 280:27–36
Matsushita T, Fujii-Taira I, Tanaka Y, Homma KJ, Natori S (2000) Male-specific IDGF, a novel gene encoding a membrane-bound extracellular signaling molecule expressed exclusively in testis of Drosophila melanogaster. J Biol Chem 275:36934–36941
McDermid HE, Duncan AM, Brasch KR, Holden JJ, Magenis E, Sheehy R, Burn J, Kardon N, Noel B, Schinzel A (1986) Characterization of the supernumerary chromosome in cat eye syndrome. Science 232:646–648
Mohamedali KA, Kurz LC, Rudolph FB (1996) Site-directed mutagenesis of active site glutamate-217 in mouse adenosine deaminase. Biochemistry 35:1672–1680
Niedzwicki JG, Abernethy DR (1991) Structure–activity relationship of ligands of human plasma adenosine deaminase2. Biochem Pharmacol 41:1615–1624
Niedzwicki JG, Liou C, Abernethy DR, Lima JE, Hoyt A, Lieberman M, Bethlenfalvay NC (1995) Adenosine deaminase isoenzymes of the opossum Didelphis virginiana: initial chromatographic and kinetic studies. Comp Biochem Physiol B Biochem Mol Biol 111:291–298
Papadakis MN, Patrinos GP (1999) Contribution of gene conversion in the evolution of the human beta-like globin gene family. Hum Genet 104:117–125
Riazi MA, Brinkman-Mills P, Nguyen T, Pan H, Phan S, Ying F, Roe BA, Tochigi J, Shimizu Y, Minoshima S, Shimizu N, Buchwald M, McDermid HE (2000) The human homolog of insect-derived growth factor, CECR1, is a candidate gene for features of cat eye syndrome. Genomics 64:277–285
Ribard C, Rochet M, Labedan B, Daignan-Fornier B, Alzari P, Scazzocchio C, Oestreicher N (2003) Sub-families of alpha/beta barrel enzymes: a new adenine deaminase family. J Mol Biol 334:1117–1131
Ribeiro JM, Charlab R, Valenzuela JG (2001) The salivary adenosine deaminase activity of the mosquitoes Culex quinquefasciatus and Aedes aegypti. J Exp Biol 204:2001–2010
Riveros-Rosas H, Julian-Sanchez A, Villalobos-Molina R, Pardo JP, Pina E (2003) Diversity, taxonomy and evolution of medium-chain dehydrogenase/reductase superfamily. Eur J Biochem 270:3309–3334
Rogozin IB, Lyons-Weiler J, Koonin EV (2000) Intron sliding in conserved gene families. Trends Genet 16:430–432
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schinzel A, Schmid W, Fraccaro M, Tiepolo L, Zuffardi O, Opitz JM, Lindsten J, Zetterqvist P, Enell H, Baccichetti C, Tenconi R, Pagon RA (1981) The “cat eye syndrome”: dicentric small marker chromosome probably derived from a no.22 (tetrasomy 22pter to q11) associated with a characteristic phenotype. Report of 11 patients and delineation of the clinical picture. Hum Genet 57:148–158
Schmitt DM, Brower DL (2001) Intron dynamics and the evolution of integrin beta-subunit genes: maintenance of an ancestral gene structure in the coral, Acropora millepora. J Mol Evol 53:703–710
Sideraki V, Wilson DK, Kurz LC, Quiocho FA, Rudolph FB (1996) Site-directed mutagenesis of histidine 238 in mouse adenosine deaminase: substitution of histidine 238 does not impede hydroxylate formation. Biochemistry 35:15019–15028
Stoltzfus A, Logsdon JMJ, Palmer JD, Doolittle WF (1997) Intron “sliding” and the diversity of intron positions. Proc Natl Acad Sci USA 94:10739–10744
Swofford DL (2001) PAUP*: Phylogenetic Analysis Using Parsimony (*and other methods) version 4.0b8. Sinauer Associates, Sunderland, MA
Taylor JS, Braasch I, Frickey T, Meyer A, Van de Peer Y (2003) Genome duplication, a trait shared by 22000 species of ray-finned fish. Genome Res 13:382–390
Tyshenko MG, Walker VK (1997) Towards a reconciliation of the introns early or late views: triosephosphate isomerase genes from insects. Biochim Biophys Acta 1353:131–136
Ungerer JP, Oosthuizen HM, Bissbort SH, Vermaak WJ (1992) Serum adenosine deaminase: isoenzymes and diagnostic application. Clin Chem 38:1322–1326
Valenzuela JG, Pham VM, Garfield MK, Francischetti IM, Ribeiro JM (2002) Toward a description of the sialome of the adult female mosquito Aedes aegypti. Insect Biochem Mol Biol 32:1101–1122
Van der Weyden MB, Kelley WN (1976) Human adenosine deaminase. Distribution and properties. J Biol Chem 251:5448–5456
Venkatesh B, Ning Y, Brenner S (1999) Late changes in spliceosomal introns define clades in vertebrate evolution. Proc Natl Acad Sci USA 96:10267–10271
Wang Z, Quiocho FA (1998) Complexes of adenosine deaminase with two potent inhibitors: X-ray structures in four independent molecules at pH of maximum activity. Biochemistry 37:8314–8324
Weijer CJ (2004) Dictyostelium morphogenesis. Curr Opin Genet Dev 14:392–398
Wilson DK, Rudolph FB, Quiocho FA (1991) Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science 252:1278–1284
Zurovec M, Dolezal T, Gazi M, Pavlova E, Bryant PJ (2002) Adenosine deaminase-related growth factors stimulate cell proliferation in Drosophila by depleting extracellular adenosine. Proc Natl Acad Sci USA 99:4403–4408
Acknowledgments
We would like to thank Fang Yang for sequencing the chicken CECR1 clone. Rezika Zurch and Twila Yobb sequenced the mouse ADAL clone. N. Ueno and H. Lillehoj provided the Xenopus and chicken cDNA clones, respectfully. We thank Isabelle Delisle for critical reading of the manuscript. Special thanks go to Warren Gallin for guidance and helpful discussions throughout the phylogenetic analysis. This study was supported by the Canadian Institutes of Health Research (H.E.M.). S.A.M. held graduate studentships from the Canadian Institutes of Health Research and the Alberta Heritage Foundation for Medical Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Martin Kreitman]
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Maier, S.A., Galellis, J.R. & McDermid, H.E. Phylogenetic Analysis Reveals a Novel Protein Family Closely Related to Adenosine Deaminase. J Mol Evol 61, 776–794 (2005). https://doi.org/10.1007/s00239-005-0046-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0046-y