
Abstract
The Pten gene was initially identified in humans as a tumor suppressor. It has since been shown to play important roles in the control of cell size, cell motility, apoptosis, and organ size, and it has also been implicated in aging. Pten is highly conserved among organisms as diverse as nematodes, insects, and vertebrates. In contrast, a phylogenetic analysis by maximum likelihood of a 133-amino acid region showed an average nonsynonymous-to-synonymous rate ratio of 10.4 for Pten in the lineage leading to parasitoid wasps of the Nasonia genus, indicating very strong positive selection. A previous study identified Pten as a potential QTL candidate gene for differences in male wing size in Nasonia. Most of the amino acid replacements that occurred in the Nasonia lineage cluster in a small region of the protein surface, suggesting that they might be involved in an interaction between Pten and another protein. The phenotypic changes due to Pten are not yet known, although it is not associated with known differences in male wing size. Introgression of Pten from one species to another does affect longevity, but a causal relationship is not established.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pten (phosphatase and tensin homologue deleted on chromosome 10) was initially identified in humans as a tumor suppressor gene (Li 1997; Steck et al. 1997) and is one of the most common targets of mutation in human cancer (reviewed in Maehama and Dixon 1999). The Drosophila homologue of the mammalian Pten gene plays a critical role in the control of cell proliferation, cell size, apoptosis, cell motility, and organ size during development (Yamada and Araki 2001). An important function of Pten appears to be as a lipid phosphatase involved in regulating important signal transduction pathways, such as the insulin, focal adhesion, and apoptosis pathways (Tamura et al. 1998; Podsypanina et al. 1999; Weng et al. 2002). Pten has also been implicated in aging, possibly through its effects on insulin signaling (Bringold and Serrano 2000). The crystal structure of Pten has been determined, and the Pten protein is composed of three primary regions, a phosphatase domain, a C2 domain, and a C terminal region (Das et al. 2003; Lee et al. 1999). Pten is evolutionary highly conserved between organisms ranging from nematodes to mammals to insects. For example, the 133-amino acid region under study here is identical between human and the mouse Mus musculus (approximately 75 million years diverged [Waterston et al. 2002]) and shows only 8 amino acid changes between human and the teleost fish Fugu rubripes (approximately 400 million years diverged [Kumar et al. 1998]).
We originally became interested in Pten because of its possible involvement in a difference in male wing size between two closely related wasp species of the Nasonia genus. Males of N. vitripennis have small vestigial forewings, whereas in N. giraulti, males have more than twofold larger wings. This size difference is due almost entirely to differences in cell size (Weston et al. 1999; J. Werren, unpublished data). One of our approaches was to study the genes of the insulin pathway because they have been identified as being important in regulating cell growth and cell size in various organisms (Coelho and Leevers 2000; Kozma and Thomas 2002). As the first step, we determined whether some of those genes were located in regions where quantitative trait loci (QTL) for wing size or shape had been mapped in the Nasonia genome (Gadau et al. 2002). Regions from five genes of the insulin signaling pathway were amplified by degenerate polymerase chain reaction and sequenced for N. vitripennis and N. giraulti. Among those, four genes showed no replacement changes between the two species. However, in the exons of the fifth gene, Pten, we observed two replacement changes but no synonymous change between N. vitripennis and N. giraulti. Furthermore, Pten was found to map to a region containing two QTLs for male wing size (J. Werren, unpublished results).
The occurrence of nonsynonymous changes and the absence of synonymous changes observed between the two Nasonia species was suggestive of positive selection. However, because of the low level of divergence between the two species, it was not possible to reach a conclusion. We have thus performed a phylogenetic analysis of Pten variation among related parasitoid wasps to determine whether there is evidence of positive selection in the lineage leading to this group of insects. We found a very high level of nonsynonymous/synonymous substitution in the lineage leading to Nasonia, demonstrating strong directional selection.
Materials and Methods
Molecular Methods
Pteromalidae is a family of parasitic wasps in the chalcidoidea. We analyzed seven species of pteromalid wasps: N. vitripennis, N. giraulti, Trichomalopsis dubius, Urolepis rufipes, Dibrachys cavus, Muscidifurax raptor, and Muscidifurax uniraptor. Genomic DNA was extracted from one haploid male with the QIAgen DNAeasy kit, following the manufacturer’s instructions (Valencia, CA). Pten amino acid sequences from Drosophila melanogaster, Anophele gambiae, Homos sapiens, Mus musculus, Fugu rubripes, and Danio rerio were retrieved from GenBank and aligned using ClustalW. The following degenerate primers were designed from conserved regions of the alignment.
-
Pten-F: 5′-AT(A/T/C)AT(A/T)GC(C/T)AT(C/G)GG(A/T/C) T(A/T)TCC(A/T)GC-3′
-
Pten-R: 5′-AA(A/T/G/C)GT(A/G)TT(A/T/G/C)AGCCA(A/G)AA(A/G)TG-3′
Genomic DNA from N. vitripennis and N. giraulti was amplified by polymerase chain reaction (PCR) at an annealing temperature of 50°C. The PCR products were directly sequenced on both strands using standard methods. PCR amplifications always produced only one band and direct sequencing always resulted in a clean chromatogram, without multiple peaks at any positions, suggesting that only one copy of Pten is present in the Nasonia genome.
The following Pten primers were designed using the following Nasonia sequences.
-
Pten-N-F: 5′-ATTACACGATTTACAATCTG-3′
-
Pten-N-R: 5′-AGAATGGCTTTATCAATTCC-3′
Due to the relatively low level of amplification achieved with the degenerate primers, for the other Pteromalid species, Pten sequences were obtained by performing a first PCR with the degenerate primers, followed by a nested PCR with the Nasonia primers. Using this procedure, we were able to amplify a 615-bp fragment of the gene consisting of two introns and two exons encoding 133 amino acids (GenBank accession numbers DQ324784 – DQ324790). Among those, 123 were from the phosphatase domain and include the phosphatase signature motif, and 10 were from the C2 domain of the protein. It should be noticed that the full PTEN protein contains 403 amino acids (Lee et al. 1999). The 133 amino acids fragment under study here thus represent about one-third of the protein. Amplifications with several degenerate primers designed to amplify other fragments of the protein in wasps were unsuccessful, possibly due to the lower conservation of the C2 domain of the protein (Goberdhan et al. 1999).
Tests of Positive Selection
We performed several analyses to determine if positive selection was involved in the evolution of Pten in Nasonia. We first used MEGA 3.0 (Kumar et al. 2004) to compare the relative abundance of synonymous and nonsynonymous substitutions between pairs of sequences. We estimated the number of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) using the method implemented in MEGA 3.0 (Kumar 2004). This method is modified from the original methods of Pamilo and Bianchi (1993), Li (1993), and Comeron (1995) and is able to handle some problematic degeneracy class assignments (Kumar et al. 2004). The variances of dS and dN were computed by bootstrap (10,000 replicates). With this information, we have tested the null hypothesis of neutral evolution (H0: dN = dS) versus the hypothesis of positive selection (H1: dN > dS) using a Z-test: Z = (dN – dS)/SQRT(Var(dS) + Var(dN)).
To test for positive selection, we also used codon-based models of sequence evolution (Yang et al. 2000). We first used likelihood ratio tests (Huelsenbeck and Rannala 1997) to determine which model of DNA sequence evolution was the most appropriate for the Pten data. We used Modeltest 3.06 (Posada and Crandall 1998) to test hierarchically the effect of unequal base frequencies, different rates between transitions and transversions, different rates between all substitutions, and rate variation over nucleotide sites. The model that best fit the dataset included unequal base frequencies and a transition/transversion rate of 5.03, i.e., a HKY85 model (Hasegawa et al. 1985). There was also significant rate heterogeneity among sites (gamma distribution with a shape parameter of 0.990). Maximum likelihood estimates of the Pten phylogeny (Fig. 1) were obtained with PAUP*4.0 (Swofford 2002) assuming this model of nucleotide substitution. Bootstrap proportions (Felsenstein 1985) were obtained from 500 pseudoreplicates. Neighbor-joining (assuming the HKY85 model of nucleotide substitution) and unweighted parsimony were also used to estimate phylogenetic relationships among the genes. In both cases, the trees had the same topology as the maximum likelihood tree shown in Fig. 1. We then used likelihood ratio tests to evaluate two codon-based models of sequence evolution, as described by Yang and coworkers (Yang 2000; Yang et al. 2000). We compared the model M0, which assumes a single ω ratio (dN/dS = ω) for all nucleotide sites and branches of the phylogeny, with a model that estimates two different ω ratios, one for the lineage of interest (“foreground lineage”) and another for all the other lineages (“background” lineages) (Yang 1998). The two-ratio model is an extension of the M0 model with one more parameter, i.e., the two models are nested. In this case, twice the difference between the likelihood of the two models will be asymptotically distributed as a chi-square random variable (Goldman and Yang 1994) with one degree of freedom (the difference in the number of parameters between the two models). The CODEML program in the PAML version 3.14 computer package (Yang 1997) was used for this analysis, assuming the tree shown in Fig. 1.

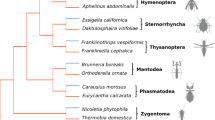
Phylogenetic relationships among Pten sequences from pteromalid wasps. The Nasonia lineage (see text) is in boldface. Tree topology was estimated using maximum likelihood (HKY85). All nodes have bootstrap values of 100% except the N. giraulti/N. vitripennis node (bootstrap value of 67%). Branch length is proportional to estimated number of nonsynonymous (A) or synonymous (B) changes. C The numbers of nonsynonymous (n) and synonymous (s) substitutions per sequence per branch are presented as n/s above each branch of the phylogeny.
Finally, we also employed the method of Zhang et al. (1998) to test for the presence of positive selection in Nasonia. Briefly, this method consists of inferring nucleotide sequences at all ancestral nodes, deducing the numbers of synonymous and nonsynonymous substitutions for each branch of the phylogeny, and then comparing these numbers with their expected values under the hypothesis of neutral evolution. To reconstruct ancestral sequences, we performed a joint reconstruction by maximum likelihood using the algorithm of Pupko et al. (2000).
Results and Discussion
We performed several analyses to determine if positive selection was involved in the evolution of Pten in Nasonia. We first used MEGA 3.0 to calculate the nonsynonymous-to-synonymous substitution rate ratio (dN/dS = ω) for all pairs of sequences by the Kumar method (Kumar et al. 2004). Because the sequences of N. giraulti, N. vitripennis, and T. dubius are very closely related, we included only the sequence of N. giraulti in this analysis. Using N. vitripennis or T. dubius produced almost-identical results. A Z-test of positive selection was performed to determine whether dN was significantly greater than dS. dN was greater than dS in 2 of 10 sequence pairs, and significantly so in 1 case (Table 1). Note, however, that the probabilities reported in Table 1 were not adjusted for multiple testing and should therefore be interpreted with caution. The high values of ω for some sequence pairs suggested that positive selection might been involved in the evolution of Pten in these Pteromalid wasps. ω was very variable between sequence pairs, with values ranging from 0 to 2.52, which suggests heterogeneity between the evolutionary lineages. We therefore used likelihood ratio test to compare codon-based models of sequence evolution, in order to determine whether selective pressures differed between the lineage of interest and the other branches of the phylogeny.
We compared the model M0, which assumes a single ω ratio (dN/dS = ω) for all nucleotide sites and branches of the phylogeny, with a model that estimates two different ω ratios, one for a “foreground lineage” and one for all the other lineages (Yang 1998). The foreground lineage needs to be a priori specified. In our case, we were interested in testing the existence of positive selection in the two Nasonia species. The foreground lineages should thus have been the terminal branches leading to these species. However, the two species are very closely related and these two branches do not show any synonymous substitutions. In order to be able to estimate the nonsynonymous/synonymous rate ratio, we therefore included in the foreground lineage the branch leading to the N. vitripennis and N. giraulti node and the branch leading to T. dubius (Fig. 1). This foreground lineage is hereafter referred to as the Nasonia lineage. The LRT statistic for the comparison of model M0 with the two-ratio model was 2 (diff. lnL) = 27.5 (df = 1; p < 0.001). This highly significantly better fit of the two-ratio model to the data indicates that the Nasonia lineage has experienced different levels of selective pressure than the other Pteromalid wasps. The estimated ω values for the two-ratio model were 0.042 for the background lineage and 10.37 for the foreground lineage. The value of ω well above 1 estimated for the Nasonia lineage indicates that it has probably experienced strong positive selection. Note that maximum likelihood methods can mistake recombination as evidence for positive selection (e.g., Anisimova et al. 2003), but as our data set consists of interspecific sequences, this is unlikely to be a problem here.
Several studies have shown that inferences of positive selection based on maximum likelihood analyses are sometimes unreliable (e.g., Suzuki and Nei 2004; Zhang 2004; but see Wong et al. 2004). Therefore, to assess the robustness of our result to the methods used, we also employed the method of Zhang et al. (1998) to test for the presence of positive selection on the Nasonia lineage. This method consists of inferring nucleotide sequences at all ancestral nodes, deducing the numbers of synonymous (s) and nonsynonymous (n) substitutions for each branch of the phylogeny and then comparing the s and n values with their expected numbers under the hypothesis of neutral evolution.
We inferred the ancestral nucleotide sequences by a maximum likelihood joint reconstruction using the algorithm of Pupko et al. (2000). The posterior probability of the inference was usually high (average value per codon of 0.995) except for one codon, number 68, at which there were two almost equally probable ancestral sequences (p = 0.57 and p = 0.42). We then calculated the numbers of nonsynonymous and synonymous substitutions for each tree branch (see Fig. 1). On the Nasonia lineage, there were 17 nonsynonymous substitutions and 0 or 1 synonymous substitution, depending on the ancestral sequence of codon number 68. To test the null hypothesis of neutral evolution (equal rates of synonymous and nonsynonymous substitutions), we estimated the numbers of potential synonymous sites (S) and potential nonsynonymous sites (N) for the sequences compared using the method of Nei and Gojobori (1986). We obtained approximately S = 93 and N = 306 for all sequences. We then applied Fisher’s exact test for examining the statistical significance of the difference between n/N and s/S in the Nasonia lineage. The test showed that the difference is significant (p = 0.006 or p = 0.012, depending on the ancestral sequence chosen at codon number 68). This result supports the fact that positive selection operated during the evolution of Pten in the Nasonia lineage.
Typically, adaptive evolution in a protein occurs at only a few sites, as most amino acids are under structural and functional constraints. Several methods have therefore been developed to test selection on individual codon sites over an entire phylogenetic tree (Nielsen and Yang 1998; Suzuki and Gojobori 1999; Yang et al. 2000). We have not used these methods here because, given the relatively low level of divergence and low number of sequences of our dataset, these analyses would have had very low power (Anisimova et al. 2002). However, we have examined the position in the protein of the 15 amino acids corresponding to the 17 nonsynonymous changes that took place in the Nasonia lineage according to our ancestral sequence reconstruction, to see if it could help us determine which selective pressure have occurred. In the region under study here, 63 amino acids are conserved among human, Drosophila, and hymenopteran (Fig. 2). None of the replacements that occurred in the Nasonia lineage belong to this category of highly conserved sites (Fig. 2), suggesting that the function of Pten is not dramatically altered in the Nasonia lineage. Mapping the observed replacements onto the three-dimensional structure of Pten (Lee et al. 1999) reveals that 11 of them occur in a small region of the protein surface (Fig. 3). This may indicate that these amino acids are involved in an interaction between PTEN and an unknown protein, though this would need to be confirmed by further experiments. The last four replacements amino acids are located at the junction between the phosphatase and the C2 domain (Fig. 3) and could potentially affect the 3D structure of the protein.

Sequence alignment of predicted partial PTEN proteins from seven Pteromalid wasps—Apis mellifera (GenBank accession no. NW_623685), Drosophila melanogaster (AAF23235), Xenopus laevis (AAD46165), Fugu rubripes (AAL08419), Mus musculus (NP_032986)—and human (AAD13528). The analyzed fragment encompasses 123 amino acids of the phosphatase domain and 10 amino acids of the C2 domain of the PTEN protein. The signature motif of protein tyrosine phosphatase, C-(X)5-R, is indicated by a line below the protein sequence. The three species that make up the Nasonia lineage (see text) are in boldface. The 15 amino acids that underwent replacements in the Nasonia lineage are shaded in gray. Note that positive selection analyses were performed on the pteromalid wasps sequences exclusively.

Three-dimensional structure of PTEN visualized with Swiss-PdbViewer (http://www.expasy.org/spdbv/). The amino acids that correspond to the nonsynonymous changes of the Nasonia lineage cluster in a small region of the protein surface (white residues at the left) or at the junction between the phosphatase and the C2 domain (light-gray residue on the center of the molecule).
The selective forces favoring the accelerated evolution of Pten in the Nasonia lineage remain undetermined. The ω values estimated by maximum likelihood represent average rates over the different branches that make up the Nasonia lineage, and they do not make it possible to pinpoint when exactly the phase of adaptive evolution occurred. By backcrossing the N. giraulti allele of Pten into N. vitripennis, we have been able to show that it is not associated with male wing size differences between the species (J. Werren, unpublished results). The three species of the Nasonia lineage differ from other Pteromalid wasps in a number of other features, including activity level, diapause tendency, male longevity and morphology. Furthermore, Pten is known to affect a range of functions, including cell proliferation, cell size, apoptosis, cell motility, organ size, and aging (Bringold and Serrano 2000; Hwangbo et al. 2004; Yamada and Araki 2001). This makes difficult to infer which selective forces operated on Pten.
Nevertheless, preliminary experiments are consistent with Pten having an influence on longevity in Nasonia. N. giraulti males have shorter lifespans than do N. vitripennis males under conditions where the males are provided with water but no nutrition. To investigate the possibility that Pten influences longevity, we introduced the Pten allele from N. giraulti (hereafter Pten-g) into an N. vitripennis background by six generations of backcrossing. Each generation, heterozygous Pten-g/Pten-v females were selected and crossed to N. vitripennis males. The alternative Pten alleles were identified by PCR-RFLP. After six generations, a homozygous Pten-g strain was established by crossing Pten-g males to Pten-g/Pten-v females and selecting for homozygous females, which were then crossed to Pten-g males. By this point, the average proportion of Ng within the genome of the backcrossed line is expected to be 3%, whereas the lines are fixed for Pten-g and tightly linked N. giraulti alleles. Males of the Pten-g strain showed significant reduction in longevity relative to the standard N. vitripennis line from which it was derived (109.7 ± 32.2 versus 144.3 ± 26.6 hr; t-test, p < 0.05).
To test for whether longevity effects are associated with Pten, Pten-g males from one line were crossed to N. vitripennis females to create heterozygous females. These were then set as virgins to produce F2 haploid males. One set was genotyped as young adults, and another provided with water until approximately 90% had died. The survivors were then genotyped. Although 43.6% of young males were Pten-g (N = 39), only 14.3% of survivors were (N = 42; χ2 = 8.55, 1 df; p < 0.01).
These results suggest that introgression of the genomic region of N. giraulti (reduced male longevity) containing Pten into N. vitripennis decreases male longevity, although it is not yet determined whether this can be attributed to Pten or flanking genes. Furthermore, it will need to be established whether this is due to an interspecies genetic incompatibility versus a specific longevity effect of Pten. Given the importance of Pten in several cellular and organismal phenotypes in diverse organisms, it is worthwhile to determine what phenotypes are affected by rapid evolution at this locus. Furthermore, the entire Pten in these lineages should now be sequenced to determine whether other portions are involved in directional selection. Examination of known interacting proteins (e.g., FAK and Shc [Yamada and Araki 2001]) could reveal whether complementary adaptive changes are occurring suggestive of specific protein-protein interactions.
For a long time, well-established cases of positive selection have remained rare. More recently, partly due to the development of more powerful statistical analyses (reviewed by Yang and Bielawski, 2000), numerous cases of positive selection have been identified. In a wide range of species, significantly elevated dN/dS ratios have frequently been described among two main gene categories: those involved in host-parasite arm race (Bishop et al. 2000; Ford 2001; Jiggins et al. 2002; Urwin et al. 2002) and those involved in reproduction (reviewed by Howard 1999; Swanson and Vacquier 2002). Though much less frequently, positive selection has also been reported in various genes, like those involved in digestion, in electron transport, or hormones (e.g., Ward et al. 1997; Goldberg et al. 2003; Wallis 2001). In primate lineages, positive selection has been reported for the genes angiogenin and BRCA1, which are involved in the regulation of cell division (Huttley et al. 2000; Zhang and Rosenberg 2002). In addition, Nielsen et al. (2005) recently found several genes involved in tumor suppression and apoptosis among those showing the strongest signs of positive selection between human and chimpanzee. Our study suggests that strong positive selection among genes involved in cell growth and proliferation might not be restricted to primates.
References
Anisimova M, Bielawski JP, Yang Z (2002) Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol 19:950–958
Anisimova M, Nielsen R, Yang Z (2003) Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164:1229–1236
Bishop JG, et al. (2000) Rapid evolution in plant chitinases: molecular targets of selection in plant-pathogen coevolution. Proc Natl Acad Sci US 97:5322–5327
Bringold F, Serrano M (2000) Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol 35:317–329
Coelho C, Leevers S (2000) Do growth and cell division rates determine cell size in multicellular organisms? J Cell Sci 113:2927–2934
Comeron JM (1995) A method for estimating the numbers of synonymous and nonsynonymous substitutions per site. J Mol Evol 41:1152–1159
Das S, Dixon JE, Cho W (2003) Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA 100:7491–7496
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Ford MJ (2001) Molecular evolution of transferrin: evidence for positive selection in salmonids. Mol Biol Evol 18:639–647
Gadau J, Page R, Werren J (2002) The genetic basis of the interspecific differences in wing size in Nasonia (Hymenoptera; Pteromalidae): major quantitative trait loci and epistasis. Genetics 161:673–684
Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C (1999) Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev 13:3244–258
Goldberg A, Wildman DE, Schmidt TR, Huttemann M, Goodman M, Weiss ML, Grossman LI (2003) Adaptive evolution of cytochrome c oxidase subunit VIII in anthropoid primates. Proc Natl Acad Sci USA 100:5873–5878
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736
Hasegawa M, Iida Y, Yano T, Takaiwa F, Iwabuchi M (1985) Phylogenetic relationships among eukaryotic kingdoms inferred from ribosomal RNA sequences. J Mol Evol 22:32–38
Howard DJ (1999) Conspecific sperm and pollen precedence and speciation. Annu Rev Ecol Syst 30:109–132
Huelsenbeck JP, Rannala B (1997) Phylogenetic methods come of age: testing hypotheses in an evolutionary context. Science 276:227–232
Huttley GA, Easteal S, Southey MC, Tesoriero A, Giles GG, McCredie MR, Hopper JL, Venter DJ (2000) Adaptive evolution of the tumour suppressor BRCA1 in humans and chimpanzees. Australian Breast Cancer Family Study. Nat Genet 25:410–413
Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M (2004) Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429:562–566
Jiggins FM, Hurst GDD, Yang Z (2002) Host-symbiont conflicts: positive selection on an outer membrane protein of parasitic but not mutualistic Rickettsiaceae. Mol Biol Evol 19:1341–1349
Kozma SC, Thomas G (2002) Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. BioEssays 24:65–71
Kumar S, Hedges SB (1998) A molecular timescale for vertebrate evolution. Nature 392:917–920
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163
Lee J, Yang H, Georgescu M, Di Cristofano A, Maehama T, Shi Y, Dixon J, Pandolfi P, Pavletich N (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99:323–334
Li WH (1993) Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 36:96–99
Li WH (1997) Molecular evolution. Sinauer Associates, Sunderland, MA
Maehama T, Dixon JE (1999) PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 9:125–128
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fledel-Alon A, Tanenbaum DM, Civello D, White TJ, J Sninsky J, Adams MD, Cargill M (2005) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3:e170
Pamilo P, Bianchi NO (1993) Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol 10:271–281
Podsypanina K, Hedrick Ellenson L, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R (1999) Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA 96:1563–1568
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Pupko T, Pe’er I, Shamir R, Graur D (2000) A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol Biol Evol 17: 890–896
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q233 that is mutated in multiple advanced cancers. Nat Genet 15:356–362
Suzuki Y, Gojobori T (1999) A method for detecting positive selection at single amino acid sites. Mol Biol Evol 16:1315–1328
Suzuki Y, Nei M (2004) False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol 21:914–921
Swanson WJ, Vacquier VD (2002) The rapid evolution of reproductive proteins. Nature Rev Gen 3:137–144
Swofford DL (2002) PAUP* Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates, Sunderland, MA
Tamura M, Gu J, Matsumoto K, Aota SI, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280:1614–1617
Urwin R, Holmes EC, Fox AJ, Derrick JP, Maiden MCJ (2002) Phylogenetic evidence for frequent positive selection and recombination in the meningococcal surface antigen PorB. Mol Biol Evol 19:1686–1694
Wallis M (2001) Episodic evolution of protein hormones in mammals. J Mol Evol 53:10–18
Ward T, Honeycutt R, Derr J (1997) Nucleotide sequence evolution at the kappa-casein locus: evidence for positive selection within the family Bovidae. Genetics 147:1863–1872
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Weng LP, Brown JL, Baker KM, Ostrowski MC, Eng C (2002) PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Hum Mol Genet 11:1687–1696
Weston R, Qureshi I, Werren JH (1999) Genetics of a morphological difference between two insect species. J Evol Biol 12:586–595
Wong WS, Yang Z, Goldman N, Nielsen R (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051
Yamada KM, Araki M (2001) Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci 114:2375–2382
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15:568–573
Yang Z (2000) Maximum likelihood estimation on large phylogenies and analysis of adaptive evolution in human influenza virus. A J Mol Evol 51:423–432
Yang Z, Bielawski JP (2000) Statistical methods for detecting molecular adaptation. TREE 15:496–503
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Swanson WJ, Vacquier VD (2000) Maximum-likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites. Mol Biol Evol 17:1446–1455
Zhang J (2004) Frequent false detection of positive selection by the likelihood method with branch-site models. Mol Biol Evol 21:1332–1339
Zhang J, Rosenberg HF (2002) Diversifying selection of the tumor-growth promoter angiogenin in primate evolution. Mol Biol Evol 19:438–445
Zhang J, Rosenberg HF, Nei M (1998) Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA 95:3708–3713
Acknowledgments
We thank F. Depaulis, C. Terzian, and two anonymous reviewers for valuable comments on the manuscript and L. Enders and J. Traggis for assistance. This research was supported by grants from the U.S. National Science Foundation (DEB 9981637) and a seed grant to J.H.W. from the University of Rochester Nathan Shock Center (NIH P30AG18254).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Willie J. Swanson]
Rights and permissions
About this article
Cite this article
Baudry, E., Desmadril, M. & Werren, J. Rapid Adaptive Evolution of the Tumor Suppressor Gene Pten in an Insect Lineage. J Mol Evol 62, 738–744 (2006). https://doi.org/10.1007/s00239-005-0002-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0002-x