
Abstract
Satellite DNAs are known for an unusual and nonuniform evolution characterized by rapid evolutionary change between species and concerted evolution leading to molecular homogeneity within species. In this paper we use satellite DNAs for phylogenetic analysis of a rapidly evolving lineage of spiders and compare the phylogeny with a hypothesis previously generated based on mitochondrial DNA and allozymes. The spiders examined include almost all species within a monophyletic clade of endemic Hawaiian Tetragnatha species, the spiny-leg clade. The phylogeny based on satellite sequences is largely congruent to those produced by mtDNA and allozymes, except that the satellite DNA yields much longer branches, with higher levels of support for any given node. Closely related species that have differentiated ecologically within an island are well resolved with satellite DNA but much less so with mtDNA. These results suggest that Tetragnatha stDNA repeats seem to be evolving gradually and cohesively during the diversification of these endemic Hawaiian spiders. The study also reveals gain–loss of satellite DNA copies during species diversification. We conclude that satellite DNA sequences may potentially be very useful for resolving relationships between rapidly evolving taxa within an adaptive radiation. In addition, satellite DNA as a nuclear marker suggests that hybridization or peripatry could play a possible role in species formation that cannot be revealed by mitochondrial markers due to its maternal inheritance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multicopy gene families such as nuclear ribosomal DNA (rDNA) exist in hundreds to thousands of copies/repeats that are reiterated tandemly at one or more chromosomal locations. Repeats generally evolve in a concerted manner leading to a complete homogenization of repeats at the intraspecific level and fixation of species-specific repeats with diagnostic mutations during species diversification (Elder and Turner 1995). The large difference in rates of segregation of chromosomes in a sexual population versus the turnover of repeats within and between chromosomes due to unequal crossing-over and/or gene conversion is the main factor driving the gradual and cohesive turnover of copies produced by mutation (molecular drive [Dover 2002]). Concerted evolution ensures that all rDNA repeats are nearly identical in sequence at the intraspecific level, within and between loci (i.e., orthologs and paralogs are identical), and hence it does not generally matter which copy is used for interspecific comparisons (Álvarez and Wendel 2003). Consequently, despite the high number of repeats, rDNA sequences generally reflect species diversification and hence have the potential to be widely used as phylogenetic markers.
Satellite DNA (stDNA) is a noncoding DNA composed of thousands to millions of tandemly arranged units that also show high intraspecific sequence identity despite their wide heterochromatic distribution. Nevertheless, the relaxed evolutionary constraints acting on stDNA monomers usually lead to marked sequence divergences at the interspecific level, rendering the signal of the satellite DNA phylogenetically uninformative (Elder and Turner 1995; Ugarkovic and Plohl 2002). In a minority of cases a stDNA family is conserved over considerable evolutionary time, evolving gradually (i.e., homogenization and fixation of mutations) and reflecting species diversification in a manner similar to rDNA markers (Arnason et al. 1992; Bachmann and Sperlich 1993; de la Herran et al. 2001). Rarely, the intermediate stages of that gradual process have been witnessed (i.e., coexistence of new and ancestral copies of a stDNA family in different stages of spreading throughout the genome [Strachan et al. 1985; Pons et al. 2002]).
Adaptive radiation on islands has been used as a model system to study stDNA evolution because species tend to be very closely related and thus are more likely to conserve sequence similarity between their repeats. Examples include Hawaiian Drosophila (Miklos and Gill 1981), Canarian darkling beetles in the genus Pimelia (Pons et al. 2002), and Hawaiian Tetragnatha spiders (Pons and Gillespie 2003). Species in the genus Tetragnatha (Araneae: Tetragnathidae) constitute the largest known radiation of spiders in the Hawaiian Islands. All species are endemic and belong to one of two major clades, the “spiny-leg” clade and a diverse web-building clade; there is also a small group of web-building species that represent a separate colonization (Gillespie et al. 1994). A previous study of Hawaiian Tetragnatha using one representative from each of the three main clades suggested that all three species share satellite sequences of common origin (Pons and Gillespie 2003). The length (183–189 bp) and nucleotide composition (58% A+T rich on average) of repeats of the three Tetragnatha species studied are very similar at the intra- and interspecific levels. However, they exhibit marked sequence divergence at the interspecific level (up to 45%) but show substantial similarity in particular DNA stretches. The results suggest a common origin of Tetragnatha repeats and an evolutionary constraint on the length and nucleotide composition at the interspecific level. At the intraspecific level, the three species show different degrees of sequence identity between repeats, probably as a result of specific historical processes. T. hawaiensis stDNA exhibits strong homogenization of the monomeric sequences, as happens in T. quasimodo, although the actual repeats are higher-order units (dimers linking two divergent subfamilies of monomers). On the other hand, T. acuta shows little homogenization of repeats due to the spreading of the three nonlinked and divergent subfamilies of repeats.
Our present study focuses on a single clade of Hawaiian Tetragnatha, the spiny-leg clade, where, because they are closely related, we expect high sequence conservation of repeats to allow us to infer phylogenetic relationships from divergence of satellites, provided the evolution of the repetitive DNAs in these spiders is dictated by “gradual” processes. Alternatively, if the repetitive DNAs evolve through “saltatory” processes such as the “library hypothesis” (Mestrovic et al. 1998; Pons et al. 2004), divergence of repeats will bear no connection to phylogenetic relationships. The phylogenetic signal of our stDNA sequences is compared to an independent and resolved phylogeny based on analysis of mitochondrial (12S, 16S, and COI sequences) and allozyme data. Results to date based on these data suggest that species on any one island are generally most closely related to each other (Gillespie 2004). The clade includes four discrete different “ecomorphs” (see Fig. 1a) which appear to have evolved independently on the different islands (Gillespie 1991, 2002), resulting in a one-to-one convergence of the same set of “ecomorph” types on each island. Across the archipelago, cladogenesis in the spiny-leg clade shows the typical pattern of lineage progression from older to younger islands (Wagner and Funk 1995; Hormiga et al. 2003). The most ancestral species, T. kauiensis and T. pilosa, are on the oldest island (Kaua’i), with subsequent sequential colonizations to Oahu (T. polychromata, T. tantalus, T. perrerai, and T. quasimodo), Maui Nui (T. waikamoi, T. brevignatha, T. restricta, and T. kamakou) and Hawaii (mostly populations from Maui) (Gillespie 2004).

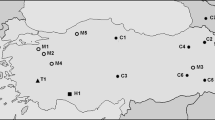
A cladogram representing the single shortest tree obtained from maximum parsimony analysis of mtDNA (12S and COI) for representatives of the spiny-leg clade of Hawaiian Tetragnatha (Gillespie et al. 1997). Numbers at each node indicate bootstrap support; numbers below branches indicate actual numbers of bases changes associated with that branch. B Rooted neighbor-joining tree of all 251 monomer sequences based on their similarity and using T. hawaiensis sequences as the outgroup. Bars represent genetic distance d = 0.05 of pairwise comparisons calculated according to the HKY+Γ model. Numbers at each node indicate bootstrap support. Bootstrap values under 50% are not indicated. The island and ecomorph are indicated for each species: green spiny (GS), green and red spiny (GRS), large brown spiny (LBS), and small brown spiny (SBS). In the center is a map of the Hawaiian Islands with the estimated ages in million years.
Materials and Methods
Sampling and Genomic DNA Isolation
Individuals from 12 endemic species of Tetragnatha “spiny-leg” spiders were collected from different localities in the Hawaiian Islands: T. quasimodo (Oahu, Mount Kaala, 600 m, and Hawaii, Puu Waawaa, 1350 m), T. perrerai (Oahu, Mount Kaala, 1350 m), T. polychromata (Oahu, Mount Kaala, 1350 m), T. tantalus (Oahu, Poamoho, 600 m), T. kamakou (Maui, Kaumakani, 1250 m), T. waikamoi (Maui, Puu Kukui, 1600 m), T. kauaiensis (Kauai, Nualolo, 1150 m), T. pilosa (Kauai, Pihea, 1200 m), T. brevignatha (Hawaii, Saddle Road, 1650 m), T. restricta (Hawaii, Hakalau, 2000 m), and T. anuenue (Hawaii, Saddle Road, 1750 m). T. hawaiensis (Kauai, Kahili, 650 m), which is also endemic to the Hawaiian Islands, was used as an outgroup. Specimens of six species (T. mohihi, T. kukuiki, T. kukuhaa, T. kikokiko, T. obscura, and T. macracantha) were not available for the current analysis. DNA was isolated from two or three frozen adult specimens stored at −80°C by standard phenol extraction and ethanol precipitation procedures.
Isolation, Cloning and Sequencing of Satellite DNA
One to two micrograms of genomic DNA was digested with restriction enzymes according to the instructions of the manufacturer (Roche, New England), and the fragments obtained were electrophoresed on 2% agarose gels. Several restriction enzymes were tested for isolation of stDNA sequences: AccI, AflII, AluI, ApaI, BfaI, BstBI, DraI, HaeIII, HincII, HinfI, MboI, NlaIV, PvuI, and SalI. Conspicuous DNA bands of about 190 and 380 bp were found. These were cut from the gel, purified using the Qiaquick Gel Extraction Kit (Qiagen), ligated into the EcoRV site of the pMOSBlue vector (Amersham Pharmacia Biotech), and used to transform Sure competent cells (Stratagene). Recombinant clones were sequenced on both strands by the dideoxy sequencing method using the Big Dye Terminator Cycle Sequencing Kit and the automatic sequencing system ABI PRISM 377 DNA Sequencer (Applied Biosystems).
Screening for the Presence of Species-Specific stDNA Sequences in Other Species of the Spiny-Leg Clade by Slot-Blot
One hundred nanograms of genomic DNA were blotted onto nylon membranes, denatured with 0.5 N NaOH, and cross-linked to the membrane (Osmotics) by exposure to ultraviolet light. Labeling of the probe, filter hybridizations, and detection of the hybridization signals were performed as described in the manual for the DIG High Prime DNA Labeling and Detection Starter Kit I (Roche). Posthybridization washes were performed allowing 60% similarity (2 × 5 min in 2 × SSC and 0.1% SDS at RT, and 2 × 15 min in 2 × SSC and 0.1% SDS at 37°C).
Sequence Analysis
We first performed multiple alignment using ClustalW v. 1.7, and aligned sequences were subjected to analysis using MODELTEST v. 3.06 (Posada and Crandall 1998) which performs a hierarchical test of likelihood (hLRT) fits under 56 different models of DNA substitution. Distance trees were built by the neighbor-joining method using the pairwise nucleotide divergences estimated using the model and parameters previously estimated with MODELTEST. Bootstrap values were calculated on 1000 replicates in PAUP*4.05 (Swofford 2002). Details of the nucleotide composition and χ2 test of homogeneity of base frequencies across sequences in the repetitive units were also estimated using PAUP* 4.05. DNA polymorphism, DNA divergence, and putative gene conversion events were estimated using DnaSP v. 3.51 (Rozas and Rozas 1997). Sequence divergence between stDNA subfamilies or species was calculated as the average nucleotide substitution per site (Dxy value from DnaSP 3.51).
Bayesian phylogenetic analyses of the Clustal alignment were performed with MrBayes 2.0 (Huelsenbeck and Ronquist 2001) using the simplest model and the HKY+Γ model selected by hLRT in MODELTEST v. 3.06. Searches were performed based on 3 million generations with four Markov chain Monte Carlo (MCMC) simulations starting from random trees which were sampled every 100 generations. The log-likelihood scores of sample points were plotted against generation time, and stationarity of Markov chains was assumed when the log-likelihood values reached a stable equilibrium (Huelsenbeck and Ronquist 2001). The stationarity of the chains was confirmed by plotting the remaining log-likelihood values (sump command). All sample points prior to stationarity were discarded as burn-in values (the first 40,000 generations), and remaining points were used to generate a strict consensus tree, with each clade posterior probability value represented by the proportion of nodes recovered in the sample of trees.
Clustal alignments were also analyzed using the parsimony criterion in PAUP* 4.05 (Swofford 2002). All nucleotide positions in the matrix were considered equivalent and weighted equally, using gaps both as missing data and as a fifth character state. Searches were performed using 1000 random additions with TBR branch swapping and the Multrees option. Bootstrap values were calculated on 100 replicates.
The separation of alignment and cladogram searching in a two-step analysis may not be the most efficient way of discovering the globally optimal solution and often leads to incompatibility of the optimal criteria applied at different stages of the analysis. Accordingly we also conducted direct character optimization analyses of the stDNA sequences as implemented in the computer program POY 2.6 (Gladstein and Wheeler 1997) because this program allows insertion and deletion events to be incorporated into the tree searches. POY implements the concept of dynamic homology with the intermediate alignment step avoided by direct assessment of the number of evolutionary events, i.e., DNA sequence transformations. The method generates more efficient explanations of sequence variation (shorter, optimal trees) than do multiple alignments and can produce multiple optimal results if more than a single optimal cladogram/alignment exists (Wheeler 1996, 2002). For the current study, the parameter space in POY was explored by varying gap costs (1, 2, and 4). For all searches reported, we present the best (lowest-cost) trees from 1000 random addition replicates and collect no more than three shortest trees in each replicate (commands: -random 1000 –maxtrees 3). Bremer support was calculated by an approximate method implemented in POY based on a search procedure using TBR swapping on a constraint file obtained with the program Jack2Hen available with the POY software. An “implied alignment” can be produced in POY which is reconstructed from the list of synapomorphies at internal nodes and is based on the initial cladogram (Wheeler 2003). The aligned matrix obtained from this procedure is a representation of the homologies established in the direct optimization and, in contrast to most standard alignment procedures, does not represent homoplastic insertions in the same column. The resulting matrix of aligned sequences is a synapomorphy scheme which approximates the character transformation in the dynamic homology search of POY (Wheeler 2003). The ClustalW alignment for the stDNA consensus sequences and the “implied alignments” for gap cost = 1, 2, and 4 are included as supplementary information.
Results
StDNA Characterization
Ten of the 11 studied species showed bands of about 190 bp after their genomic DNA was digested with restriction enzymes and analyzed by agarose gel electrophoresis (Table 1). The majority of species displayed a prominent band (weaker in T. tantalus and T. polychromata). T. anuenue genomic DNA revealed no visible bands despite having been screened with 20 different endonucleases. Some species showed a dim ladder of oligomers of the 190-bp band such as those described elsewhere in T. quasimodo and T. hawaiensis (Pons and Gillespie 2003). We cloned the monomeric bands of these 10 species and then randomly sequenced from 12 to 19 positive clones for each species for both strands (Table 1). The genomic DNA digestions of T. kauaiensis and T. kamakou also induced conspicuous dimeric bands that could be cloned to analyze the variability of contiguous repeat units. We were able to sequence two dimers from T. kauaiensis (pdKIS) and eight from T. kamakou (pdKAM). In the analysis of the repeat unit sequences we also included those isolated from T. quasimodo and T. hawaiensis (Pons and Gillespie 2003). As in T. quasimodo and T. hawaiensis, in both T. kauainesis and T. kamakou dimers the repeat unit exhibiting the restriction site BfaI was labeled pdKIS A and pdKAM A, respectively, and the unit lacking this site, pdKIS B and pdKAM B, respectively. Species from the spiny-leg clade have consensus repetitive sequences of similar length (188–191 bp). The consensus repeat unit from the outgroup species, T. hawaiensis, was shorter (183 bp). Some repeat units are shorter due to the partial deletion of the sequence: pWAI04, pWAI14, pKISO8, pKISI5, pKIS17, and pBRE16H. All Tetragnatha repeat units show similar nucleotide composition (57.99% A + T on average; p < 0.999).
Phylogenetic Analyses of stDNA Sequences Using Clustal Alignments
Tetragnatha repetitive sequences still display similarity despite their high divergence (Table 2; alignment ALIGN_000247, accession numbers AJ413315–AJ413559 and AJ413577–AJ413582). Phylogenetic analysis of all 251 repeat unit sequences based on their similarity (HKY+Γ model), using neighbor-joining with T. hawaiensis as the outgroup, placed T. pilosa and T. kauaiensis as monophyletic and basal (Fig. 1B). These sequences are the least divergent relative to the outgroup sequences, T. hawaiensis (70–65% similarity). The tree displays two other groups, both monophyletic relative to the basal sequences: (1) sequences from Oahu (T. tantalus, T. polychromata, T. perreirai, and T. quasimodo from Oahu and Hawaii) and (2) sequences from Maui and Hawaii (T. kamakou, T. waikamoi, T. brevignatha, and T. restricta). Surprisingly, T. quasimodo from Hawaii only shows one of the stDNA variants found in T. quasimodo from Oahu. The cross-hybridization of the stDNAs of each species against the genomic DNA of the other taxon recovered the same groups of species as the neighbor-joining analysis based on sequence divergence (Fig. 2). Interestingly, T. anuenue genomic DNA, in which we could not isolate any repetitive sequences, showed no hybridization with any of the stDNA sequences isolated in the other Tetragnatha species studied, even under low stringency conditions. The neighbor-joining tree revealed that, except for T. waikamoi, the repeat units have a monophyletic origin at the intraspecific level. We also performed Bayesian and parsimony searches on Clustal alignments. Since satellites were monophyletic and very similar at the intraspecific level, we used the 50% majority rule consensus sequence obtained from the repeats for each species. In those taxa showing two divergent subfamilies of monomers, T. quasimodo Oahu, T. brevignatha, and T. waikamoi, we built the 50% majority rule consensus of each subfamily to assess their monophyly. Both Bayesian and parsimony analyses of the 15 consensus sequences retrieved the same tree topology (Fig. 3A) as the neighbor-joining tree based on the 251 sequences (Fig. 1B).

Summary of the pairwise cross-hybridization results for the stDNA sequences isolated in each species (rows) with 100 ng of genomic DNA of the 12 Tetragnatha species of the spiny-leg clade included in this study plus the outgroup T. hawaiensis (columns). Hybridizations were performed allowing 60% similarity. The particular cloned unit used as probe is indicated in each case. The codes for species sequences (top row) are indicated in Table 1. Dim hybridizations are indicated by +d and strong ones by +s. Groups showing similar sequences are highlighted with different boxes. Since T. waikamoi has two very divergent subfamilies, one representative of each subfamily was used as probe.

A Phylogram representing tree obtained from a ClustalW alignment of the consensus stDNA sequences of Tetragnatha species of the spiny-leg clade (201 characters) using Bayesian inference with a HYK+Γ model. The 50% majority rule consensus sequence for each species was obtained from all clones. In those species showing two subfamilies of monomers, T. quasimodo, T. brevignatha, and T. waikamoi, we used the 50% majority rule consensus of each subfamily to assess the monophyly of those subfamilies. Parsimony analysis using gaps as missing data (28 constant characters and 149 informative ones for a tree length of 447 steps) or fifth state (19 constant characters and 156 informative ones for a tree length of 488 steps) retrieved identical topologies. Numbers above branches refer to Bremer support values (parsimony gaps missing data/parsimony gaps fifth state), and numbers below branches are bootstrap support values (parsimony gaps missing data/ parsimony gaps fifth state/ Bayesian posterior probability). B Cladogram (449 steps) obtained from same consensus sequences under gap cost = 1 using POY version 2.6. Gap cost = 2 retrieved the same topology and gap cost = 4 differed at two nodes. *T. pilosa and T. kauaiensis are monophyletic. **T. restricta is basal to T. kamakou, T. waikamoi, and T. brevighatha. Numbers at each node indicate Bremer support values for the different gap costs (1/2/4).
Phylogenetic Analyses Using Direct Optimization
Since POY requires high computational power, searches were again performed with the 15 consensus sequences. Searches were initially performed under equal weighting of all characters (-gap 1 -change 1), but alignment parameters were varied to explore the effects on topology. Under equal weighting (Fig. 3B), the topology retrieved by the POY search was very similar to that obtained in all the different analyses performed with the Clustal alignments (Figs. 1B and 3A), except that the Kauai clade (T. pilosa + T. kauaiensis) was paraphyletic. Increasing the gap cost = 2 relative to nucleotide changes had no effect on tree topology and cost = 4 had little effect. The discrepancies were the basal position of T. restricta within the clade composed of sequences from T. kamakou, T. brevignatha, and T. waikamoi and the monophyly of basal sequences from Kauai (T. pilosa + T. kauaiensis). Interestingly, total Bremer support values decreased (132, 97, 92, see Fig. 3B, for specific values for each node) as the gap opening cost increased (1, 2, and 4, respectively). Plots of uncorrected distances against total numbers of transitions or transversions, using both the Clustal alignment (see Fig. 4) and the “implied alignment” from POY (gap cost = 1; not shown), displayed no evidence of saturation in the stDNA sequences. The slope and R2 values for the “implied alignment” plot were very similar to those shown in the Clustal alignment plot (Fig. 4).

Plots of total number of transitions (black diamonds) and transversions (gray squares) as a function of uncorrected divergence in Tetragnatha stDNA sequences.
Analysis of Nucleotide Variation
The presence of two nonmonophyletic subfamilies explains the higher nucleotide diversity of T. waikamoi repetitive sequences (16.2 %). These subfamilies (WAI 1 and WAI 2) differ in length (188 and 191 bp) and by 49 fixed nucleotide positions (38.5% divergence). The nucleotide diversity within these subfamilies (3.9% and 1.7%, respectively) is much lower than the divergence between subfamilies. The variability within subfamilies is similar to the nucleotide diversity estimated in the other species (Table 1) and the divergence of T. waikamoi subfamilies is similar to the interspecific divergence of stDNA sequences (Table 2). Nucleotide diversity in other species was low (Table 1). The intraspecific polymorphism of repetitive sequences appears to be due to single nucleotide substitutions that are distributed throughout the monomer. The nucleotide substitutions are mainly autapomorphies (unique) or synapomorphies shared by few monomers. These species, as in T. hawaiensis, showed no divergent subfamilies. The exception was T. brevignatha, which possessed two monopheletic subfamilies (BRE 1 and BRE 2) supported by high bootstrap and ca. 10% divergence as in T. quasimodo from Oahu. T. brevignatha subfamilies differ in length (188 and 191 bp, respectively; positions 155, 156, and 168) and by 17 fixed nucleotide substitutions. T. brevignatha sequences showed low nucleotide diversity within variants (1.4% and 0.8%, respectively) and higher divergence between them (11.6%), like those from T. quasimodo Oahu. The analysis of the two variants of T. brevignatha reveals the presence of one sequence with a tract that has diagnostic mutations of the other variant (pBRE 05 positions 61–73), possibly caused by gene conversion. Other species such as T. kamakou, T. tantalus, and T. quasimodo Hawaii have variants differing in length (one or two indels) but neither show fixed nucleotide substitutions nor are supported by bootstrap values.
At the interspecific level some closely related species show the coexistence of ancestral and recent copies (i.e., intermediate stages of a gradual and continuous turnover of repeats in a multigene family through molecular drive [Dover 2002; Pons et al. 2002; Strachan et al. 1985]). Several positions indicating the coexistence of two major type of repeats in different stages of spreading (i.e., shared synapomorphies) were found in the closely related species T. polychromata, T. tantalus, and T. perrerai (e.g., position 62 indel/A variants, 130 T/C, 151 indel/G, 169 T/G, and 177 T/A). Similar results were found in other closely related species: T. kauaiensis and T. pilosa (position 12 T/G variants, 52 A/G, and 110 G/A), T. kamakou and T. restricta (1 G/A, 37 T/A, 139 T/A, and 140 T/A), and T. waikamoi, T. restricta, and T. brevignatha (61 C/G).
To assess the relative usefulness of stDNA versus mtDNA in phylogenetic estimation we plotted the percentage nucleotide substitution change in stDNA versus cytochrome oxidase I mtDNA for pairwise comparisons (Fig. 5). As can be seen, the rate of change of stDNA is similar to that of mtDNA at very low levels of divergence. Above 9% mtDNA sequence divergence, stDNA divergence is approximately four times higher than that of mtDNA and increases linearly with mtDNA divergence (R2 = 0.220, p = 0.001).

Relationship between percentage sequence change in satellite DNA (from Table 2) and percentage sequence change in mitochondrial DNA. For the mtDNA, estimates are based on 560 base pairs of cytochrome oxidase (Gillespie 1999). The line indicates the expected level of base frequency change in stDNA if the rates were similar to those of mtDNA. There are only three points at very low mtDNA divergences at which the rates are comparable. At mtDNA divergence levels of 5–10%, stDNA divergences are approximately four times greater than those of mtDNA.
Discussion
Tetragnatha stDNA sequences show large divergences between certain species (up to 60% in the most distant cases), which could raise some questions about the homology of the satellites. We assume that if Tetragnatha repeats undergo concerted evolution (molecular drive [Dover 2002]), making copies identical within species and thereby removing the possibly confounding effect of using multiple paralogous copies, then they should retrieve the same species diversification pattern (i.e., tree topology) as the unambiguous mitochondrial genes and allozymes. Despite the large sequence divergences, the phylogeny of the spiny-leg Tetragnatha species based on stDNA retrieves strong phylogenetic signal that is mostly concordant with that based on classical mtDNA sequences. Therefore, we assume that Tetragnatha repeats evolve in a gradual and cohesive manner, fixing species-specific mutations during species formation. Interestingly, the methods of tree building, i.e., a two-step protocol versus direct optimization, and different parameters used in the tree searches had little impact on the tree topology, indicating the unambiguous phylogenetic signal from the stDNA sequences.
The difficulty in establishing sequence identities in stDNA sequences at the interspecific level has prohibited the study of their evolutionary dynamics and homology. Despite some difficulties (e.g., failure to detect stDNA sequences in T. anuenue and the presence of two divergent variants/paralogs coexisting in T. waikamoi), the present study and other similar ones in insects (e.g., Strachan et al. 1985; Pons et al. 2002) illustrate the conditions under which an evolutionary model can be postulated (gradual evolution through molecular drive). This gradual evolutionary change has been detected over a limited evolutionary period only (usually quite short, <5 million years). However, it may occur over longer periods of time but cannot be detected due to the absence of sequence similarity, which also precludes phylogenetic analyses to assess homology and evolutionary dynamics.
Previous studies on Hawaiian Tetragnatha (Pons and Gillespie 2003) have suggested that the satellite sequences have a common origin and selective constraints act on the length and nucleotide composition of repeats at the interspecific level (Pons and Gillespie 2003). The results of the current study support the existence of such constraints. A similar pattern has been also described in Canarian Pimelia (Pons et al. 2002), but in these beetles constraints seem to act on runs composed of three or more adenines. The more relaxed selective pressure on the primary sequence of Tetragnatha repeats compared to Pimelia, in which conservation of A–T runs is inextricably linked to primary sequence conservation, could explain the lower sequence identities found in the spider repeats.
The phylogeny of the spiny-leg Tetragnatha species based on stDNA sequences and on classical mtDNA sequences is consistent with the progression rule of colonization from the oldest island, Kauai, to Oahu and down to Maui Nui (Molokai, Lanai, and Maui) and Hawaii. Both mtDNA and stDNA place T. pilosa and T. kauaiensis (Kauai) as basal species. mtDNA suggests that colonization from Kauai to Oahu gave rise to the lineage (O) that includes T. perreirai, T. polychromata, and T. tantalus (see Fig. 1). Then from this group, two sister lineages arose, the first (Q) including T. quasimodo, which has colonized each island as it appeared. The second lineage (M) also shows a pattern of progression, but with multiple ecomorphs forming independently on Maui Nui: T. waikamoi, T. restricta, and T. kamakou; and T. brevignatha and T. macracantha (Gillespie et al. 1997; Gillespie 2004). The stDNA phylogeny suggests a similar scenario, although this nuclear marker retrieves an alternative hypothesis where the main lineages (O + Q + M) may have originated on Kauai and then colonized the other islands sequentially. Interestingly, the stDNA sequences in the current study gave a similar phylogenetic signal to the mtDNA markers. Moreover, the branch lengths and support for the overall tree topology are much higher for the stDNA than for the mtDNA. The reason for this is clearly the tendency for stDNA to show high levels of intraspecific conservatism (due to concerted evolution) yet equally high levels of interspecific disparity between closely related species. This phenomenon is illustrated in Fig. 5, which shows the relationship between percentage sequence divergence in stDNA versus COI mtDNA. The bimodality in divergence rates of stDNA sequences and the linear relationship with divergence in mtDNA sequences indicate that stDNA can be useful as a marker for identifying nonrecombining gene pools and for elucidating phylogenetic relationships of very closely related species such as those that have undergone recent adaptive radiation.
The detection of intermediate stages of turnover of repeats (i.e., coexistence of older and more recent stDNA copies) and gene conversion events (a molecular mechanism driving concerted evolution) also suggests “gradual” as opposed to “saltatory” evolution (Dover 2002; Pons et al. 2002; Bachmann and Sperlich 1993; Strachan et al. 1985). However, in contrast to the rather constant fixation rate of nucleotide substitutions found in the closely related Iberian Pimelia beetles (Pons et al. 2002), Hawaiian Tetragnatha spiders show a nonconstant rate. For instance, very closely related species, such as T. polychromata and T. tantalus, have identical sequences with no species-specific fixed mutations. But other closely related species pairs such as T. kamakou and T. restricta exhibit very divergent sequence repeats (35% divergence), although their units still share few synapomorphies (i.e., mutations present in the common ancestor). This effect can also be deduced from Fig. 5, which shows that species mostly exhibit stDNA divergence of either <10% or >30%, with very little in the 10–30% range. Interestingly, complete replacement of repeat units, sometimes with very divergent repeats (e.g., T. restricta), can be achieved over short evolutionary periods since the species of Tetragnatha endemic to Maui and Hawaii cannot be older than the age of the islands (Maui ranges from 0.8 to 1.3 million years; Hawaii from 0 to 0.4 million year [Carson and Clague 1995]). Similar nonconstant rates have been described in a recent study comparing distant Pimelia species (Pons et al. 2004). However, it is unclear which particular factors are involved in this accelerated and static evolution of stDNAs in each case. Accelerated evolution of satellite nucleotide sequences might occur, for instance, in response to various forms of stress and/or retrotransposon-mediated adaptive evolution (e.g., Kalendar et al. 2000). On the other hand, biased gene conversion can lead to extreme sequence conservation if ancestral sequence variants are favored over recently generated ones during concerted evolution (Hillis et al. 1991).
Three spiny-leg species (T. waikamoi, T. brevignatha, and T. quasimodo Oahu), as well as the previously described T. acuta (Pons and Gillespie 2003), harbor divergent subfamilies of repeats in their respective genomes. The evidence of high nucleotide identity within subfamilies suggests that strong homogenization also acts at this level. The presence of two variants in T. brevignatha and in T. quasimodo imposes no difficulty for phylogenetic analysis because the variants occur at the terminal branches. In the case of T. waikamoi, variant divergence is higher than some interspecific divergence values. One explanation for this is that historical processes occurring during speciation, such as hybridization (which is so common among island radiations. [e.g., Jordan et al. 2003; Álvarez and Wendel 2003]), could have given rise to the different variants. Alternatively, the two variants may be maintained in a large population of T. waikamoi, with one giving rise to T. brevignatha and T. macracantha and the other to T. restricta and T. kamakou. Preliminary results from ITS-2 (unpublished data) also show the coexistence within a species of different variants of ITS-2, suggesting similar evolutionary patterns. On the other hand, biases in any of the multiple factors involved in the concerted evolution, such as drastic changes in population size (Ohta and Dover 1984) or a lack of nonreciprocal transfer between nonhomologous chromosomes (Trick and Dover 1984), could cause the formation and fixation of variants within species. However, such biases are an unlikely explanation for the presence of the different variants in the current study, as it would be expected to lead to incongruent phylogenetic signal between satellite and mtDNA, which we do not find.
The results presented here also suggest a scenario of gain–loss of stDNA copies through the evolutionary history of the Tetragnatha species in the spiny-leg clade, as has been described in other studies of closely related species (e.g., Panzera et al. 1995). The cross-hybridization experiments, carried out with the same amount of genomic DNA for each species, resulted in a weaker signal from the genomic DNA of T. tantalus and T. polychromota compared to T. perreirai even using its own stDNA sequences as probes, suggesting a loss of repeats. Moreover, the loss of DNA sequences and weaker hybridization in Hawaii relative to Oahu populations of T. quasimodo could be related to the absence of variant 2 of Oahu in Hawaii T. quasimodo. The impossibility of isolating stDNA sequences in another “small spiny-leg” species, T. anuenue, together with the fact that no signal was obtained in cross-hybridization experiments, suggests a complete loss of the stDNA, or enough of a loss as to make the copies undetectable by standard methods. The loss–gain and/or redistribution of heterochromatin, mainly composed of stDNA repeats, has been linked, as cause or consequence, with chromosome rearrangement and evolution (i.e., genome reorganization) and, therefore, with speciation (Brutlag 1980).
In summary, the stDNA sequences of the Tetragnatha spiny-leg spiders studied, despite their high sequence divergence, give a phylogenetic that is related to but much higher than that of mtDNA within the range of approximately 5–10% mtDNA sequence divergence (Fig. 5). The phylogenetic relationships recovered from stDNA are therefore very similar to those recovered from traditional makers but tend to accentuate differences between segregated gene pools. Both phylogenies are compatible with the geological history of the Hawaiian Islands, suggesting sequential colonization from older to newer islands. Moreover, the repetitive DNA may reflect historical processes that have occurred during speciation, such as peripatry and hybridization, which are not usually reflected by mitochondrial phylogenies but are commonly recovered in nuclear phylogenies. Given the notoriously problematic nature of phylogenetic reconstruction among species showing explosive diversification, stDNA demands some attention for its potential usefulness in these cases.
References
I A’lvarez JF Wendel (2003) ArticleTitleRibosomal ITS sequences and plant phylogenetic inference Mol Phylogenet Evol 29 417–434
U Arnason S Gretarsdottir B Widegren (1992) ArticleTitleMysticetae (baleen whale) relationships based upon the sequence of the common cetacean DNA satellite Mol Biol Evol 9 1018–1028
L Bachmann D Sperlich (1993) ArticleTitleGradual evolution of a specific satellite DNA family in Drosophila ambigua, D. tristis, and D. obscura Mol Biol Evol 10 647–659
DL Brutlag (1980) ArticleTitleMolecular arrangement and evolution of heterochromatin DNA Annu Rev Genet 14 121–144
HL Carson DA Clague (1995) Geology and biogeography of the Hawaiian Islands WL Wagner V Funk (Eds) Hawaiian biogeography: Evolution on a hot spot archipelago Smithsonian Institution Press Washington, DC 14–29
R la Herran Particlede CR Rejon MR Rejon MA Garrido-Ramos (2001) ArticleTitleThe molecular phylogeny of the Sparidae (Pisces, Perciformes) based on two satellite DNA families Heredity 87 691–697
GA Dover (2002) ArticleTitleMolecular drive Trends Genet 18 587–589
JF Elder BJ Turner (1995) ArticleTitleConcerted evolution of repetitive DNA sequences in eukaryotes Q Rev Biol 70 297–323 Occurrence Handle10.1086/419073 Occurrence Handle1:CAS:528:DyaK2sXksFGrurY%3D Occurrence Handle7568673
RG Gillespie (1991) ArticleTitleHawaiian spiders of the genus Tetragnatha. I. Spiny leg clade J Arachnol 19 174–209
RG Gillespie (1999) ArticleTitleComparison of rates of speciation in web-building and non-web-building groups within a Hawaiian spider radiation J Arachnol 27 79–85
RG Gillespie (2002) ArticleTitleHawaiian spiders of the genus Tetragnatha. IV: New, small species in the spiny leg clade J Arachnol 30 159–172
R Gillespie (2004) ArticleTitleCommunity assembly through adaptive radiation in Hawaiian spiders Science 303 356–359
RG Gillespie SR Palumbi HB Groom (1994) ArticleTitleMultiple origins of a spider radiation in Hawaii Proc Natl Acad Sci USA 91 2290–2294
RG Gillespie HB Croom GL Hasty (1997) ArticleTitlePhylogenetic relationships and adaptive shifts among major clades of Tetragnatha spiders (Araneae: Tetragnathidae) in Hawai’i Pacif Sci 51 380–394
DS Gladstein WC Wheeler (1997) POY: The optimization of alignment characters American Museum of Natural History New York
DM Hillis C Moritz CA Porter RJ Baker (1991) ArticleTitleEvidence for biased gene conversion in concerted evolution of ribosomal DNA Science 251 308–310
G Hormiga MA Arnedo RG Gillespie (2003) ArticleTitleSpeciation on a conveyor belt: sequential colonization of the Hawaiian Islands by Orsonwelles spiders (Araneae: Linyphiidae) Syst Biol 52 70–88
JP Huelsenbeck F Ronquist (2001) ArticleTitleMRBAYES: Bayesian inference of phylogenetic trees Bioinformatics 17 754–755 Occurrence Handle10.1093/bioinformatics/17.8.754 Occurrence Handle1:STN:280:DC%2BD3MvotV2isw%3D%3D Occurrence Handle11524383
S Jordan C Simon D Polhemus (2003) ArticleTitleMolecular systematics and adaptive radiation of Hawaii’s endemic damselfly genus Megalagrion (Odonata: Coenagrionidae) Syst Biol 52 89–109
R Kalendar J Tanskanen S Immonen E Nevo AH Schulman (2000) ArticleTitleGenome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence Proc Natl Acad Sci USA 97 6603–6607 Occurrence Handle10.1073/pnas.110587497 Occurrence Handle1:CAS:528:DC%2BD3cXktFaju7Y%3D Occurrence Handle10823912
N Mestrovic M Plohl B Mravinac D Ugarkovic (1998) ArticleTitleEvolution of satellite DNAs from the genus Palorus—Experimental evidence for the “library” hypothesis Mol Biol Evol 15 1062–1068
GL Miklos AC Gill (1981) ArticleTitleThe DNA sequences of cloned complex satellite DNAs from Hawaiian Drosophila and their bearing on satellite DNA sequence conservation Chromosoma 82 409–427
T Ohta GA Dover (1984) ArticleTitleThe cohesive population genetics of molecular drive Genetics 108 501–521
F Panzera R Perez Y Panzera F A’lvarez E Scvortzoff R Salvatella (1995) ArticleTitleKaryotype evolution in holocentric chromosomes of three related species of triatomines (Hemiptera–Reduviidae) Chromosome Res 3 143–150
J Pons RG Gillespie (2003) ArticleTitleCommon origin of the satellite DNAs of Hawaiian spiders of the genus Tetragnatha: Evolutionary constraints on the length and nucleotide composition of the repeats Gene 313 169–177
J Pons E Petitpierre C Juan (2002) ArticleTitleEvolutionary dynamics of satellite DNA family PIM357 in species of the genus Pimelia (Tenebrionidae, Coleoptera) Mol Biol Evol 19 1329–1340
J Pons B Bruvo E Petitpierre M Plohl D Ugarkovic C Juan (2004) ArticleTitleComplex structural features of satellite DNA sequences in the genus Pimelia (Coleoptera: Tenebrionidae): Random differential amplification from a common satellite DNA library Heredity 92 418–427
D Posada KA Crandall (1998) ArticleTitleModeltest: testing the model of DNA substitution Bioinformatics 14 817–818 Occurrence Handle10.1093/bioinformatics/14.9.817 Occurrence Handle1:CAS:528:DyaK1MXktlCltw%3D%3D Occurrence Handle9918953
J Rozas R Rozas (1997) ArticleTitleDnaSP version 2.0, a novel software package for extensive molecular population genetic analysis Comput Applic Biosci 13 307–311
T Strachan D Webb GA Dover (1985) ArticleTitleTransition stages of molecular drive in multiple-copy DNA families in Drosophila EMBO J 4 1701–1708
DL Swofford (2002) PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods) Sinauer Associates Sunderland, MA
M Trick GA Dover (1984) ArticleTitleUnexpectedly slow homogenization within a repetitive DNA family shared between two subspecies of tsetse fly J Mol Evol 20 322–329
D Ugarkovic M Plohl (2002) ArticleTitleVariation in satellite DNA profiles—causes and effects EMBO J 21 5955–5959 Occurrence Handle10.1093/emboj/cdf612 Occurrence Handle1:CAS:528:DC%2BD38XptFOrt7w%3D Occurrence Handle12426367
WL Wagner V Funk (1995) Hawaiian biogeography: Evolution on a hot spot archipelago Smithsonian Institution Press Washington, DC
WC Wheeler (1996) ArticleTitleOptimization alignment: The end of multiple sequence alignment in phylogenetics? Cladistics 12 1–9
WC Wheeler (2002) Optimization alignment: down, up, error, and improvements R DeSalle G Giribet WC Wheeler (Eds) Techniques in molecular systematics and evolution Birkhäuser Basel 55–69
WC Wheeler (2003) ArticleTitleImplied alignment: A synapomorphy-based multiple-sequence alignment method and its use in cladogram search Cladistics 19 261–268
Acknowledgments
This work was funded by a grant from the Obra Cultural i Social Sa Nostra Fundation (Spain) to J.P., with additional funds from the Schlinger Foundation, and a National Science Foundation (DEB 9726573) grant to R.G.G. For help with fieldwork, we are indebted to Haleakala National Park, the Nature Conservancy of Hawaii, Maui Pineapple Company, the State Department of Land and Natural Resources, and the Hawaii Natural Areas Reserve System. For help with collecting we owe particular thanks to M.A. Arnedo, R. Bartlett, C.F. Davenport, J. Giffin, A.C. Medeiros, D. Preston, and G.K. Roderick. We also thank M.A. Arnedo for help with the POY analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Rafael Zardoya
Appendix
Rights and permissions
About this article
Cite this article
Pons, J., Gillespie, R.G. Evolution of Satellite DNAs in a Radiation of Endemic Hawaiian Spiders: Does Concerted Evolution of Highly Repetitive Sequences Reflect Evolutionary History?. J Mol Evol 59, 632–641 (2004). https://doi.org/10.1007/s00239-004-2655-2
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2655-2