
Abstract
Since the early days of the discovery of the genetic code nonrandom patterns have been searched for in the code in the hope of providing information about its origin and early evolution. Here we present a new classification scheme of the genetic code that is based on a binary representation of the purines and pyrimidines. This scheme reveals known patterns more clearly than the common one, for instance, the classification of strong, mixed, and weak codons as well as the ordering of codon families. Furthermore, new patterns have been found that have not been described before: Nearly all quantitative amino acid properties, such as Woese’s polarity and the specific volume, show a perfect correlation to Lagerkvist’s codon–anticodon binding strength. Our new scheme leads to new ideas about the evolution of the genetic code. It is hypothesized that it started with a binary doublet code and developed via a quaternary doublet code into the contemporary triplet code. Furthermore, arguments are presented against suggestions that a “simpler” code, where only the midbase was informational, was at the origin of the genetic code.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crick (1968) introduced the notion that the genetic code is simply the result of pure chance or a “frozen accident” and that it therefore does not need any further evolutionary explanation. Later, this view was questioned. Although certain knowledge of the origin and early stages of life is not likely to be obtained, there are some hints of possible evolutionary scenarios of the genetic code. One direction of research (the “top-down approach” [Szathmary 1999]) analyzes patterns in the contemporary code (Knight and Landweber 1998; Szathmary 1999) and tries to infer appropriate chemical and selective forces. The bottom-up approach, on the other hand, is rooted in biochemistry and aims at constructing plausible scenarios for the origin of coding (Topal and Fresco 1976; Maizels and Weiner 1987; Szathmary 1993).
It has been appreciated for a long time that the genetic code assigns similar amino acids to similar codons (Sonneborn 1965; Woese 1965; Zuckerkandl and Pauling 1965; Crick 1968). Two different rationales have been presented: first, mutation (Sonneborn 1965; Zuckerkandl and Pauling 1965) and translation (Woese 1967; Haig and Hurst 1991; Freeland and Hurst 1998) error minimization (or both (Ardell and Sella 2002)) and, second, the tendency of similar amino acids to directly interact with similar RNA sequences (Woese et al. 1966; Yarus 1998, 2000). Landweber and coworkers found further evidence to support both hypotheses. Extending previous work (Haig and Hurst 1991; Freeland and Hurst 1998) by quantifying amino acid similarity, these authors were able to show that “the canonical code is at or very close to a global optimum for error minimization” (Freeland et al. 2000). Based on the earlier work of Yarus (cf. Yarus 1998, 2000), by doing a statistical analysis of RNA aptamers (nucleic acid molecules selected to bind specific ligands), they concluded that there is “the strongest support for an intrinsic affinity between any amino acid and its codons” (Knight and Landweber 1998). It has also been proposed that instead of the actual codons, some derivatives of them, such as the anticodons (Dunnill 1966; Jungck 1978) or codon–anticodon duplexes (Alberti 1997), were the original amino acid binding motifs. It could also be that the original amino acid recognition took place at the tRNA acceptor stem (Hopfield 1978) or that the specificity of aminoacylation is determined by the interaction of the tRNA synthetase with its tRNA (Weiner and Maizels 1987). Szathmary (1999) proposed that amino acid RNA allocation took place even before the appearance of tRNA. He also gave a possible evolutionary scenario for the development of an anticodon hairpin to a longer structure with an operational code at the acceptor stem.
Several patterns of the genetic code have been identified, which can be illustrated within the classical scheme.
The Common Scheme of the Genetic Code
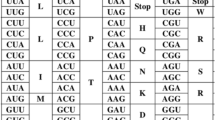
The common scheme of the genetic code (Alberts et al. 2002) contains 43 = 64 codons, a three-dimensional matrix where each dimension represents one of the three positions in the triplet code (Fig. 1). Viewed this way, some patterns emerge: The first codon position seems to be correlated with amino acid biosynthetic pathways (Wong 1975; Taylor and Coates 1989) and with their evolution as evaluated by synthetic “primordial soup” experiments (Eigen 1977; Schwemmler 1994). The second position is correlated with the hydropathic properties of the amino acids (Crick 1968; Wolfenden et al. 1979; Taylor and Coates 1989), and the degeneracy of the third position could be related to the molecular weight or size of the amino acids (Hasegawa and Miyata 1980; Taylor and Coates 1989).

The common presentation of the standard (“universal”) genetic code. All deviations from this code (Elzanowski and Ostell 2000) are thought to be the result of later mutations (Osawa et al. 1992; Knight and Landweber 2000b; Knight et al. 2001). Shaded regions show codon families.
Lagerkvist (1978, 1981) divided the common illustration scheme (Fig. 1) into a left part (containing the first and second columns, i.e., U and C in the second position of the codon, respectively) and a right part (the third and fourth columns, i.e., A and G in the second position). He observed that codon families (the amino acid of a codon family is uniquely determined by the first two nucleotides of a codon; cf. shaded regions in Fig. 1) have a much higher probability to appear in the left part. Furthermore, he found that “strong” codons (the first two nucleotides in the codon are G and/or C) always represent codon families, while “weak” codons (A and/or U as the first two nucleotides) never do so. “Mixed” codons in the right part of the scheme never represent codon families, whereas mixed codons in the left part always stand for a codon family. Lagerkvist (1978) speculated “that interactions between mixed codons and their anticodons are stronger in the left half of the codon square.”
However, most amino acid properties show no clear pattern in the common scheme of the genetic code. Instead Jungck (1978) used 15 different quantitative measures of amino acid properties such as polarity and molecular volume to demonstrate that these properties are generally more closely correlated with anticodon than with codon dinucleoside monophosphate properties. This supports the hypothesis that the relationship between amino acids and their anticodon dinucleosides was the basis for the origin of the genetic code.
In this article we follow the “top-down approach” toward understanding the organization of the genetic code. We are thereby led to propose a new classification scheme for the code that helps us to identify new patterns which in turn suggest new speculations about its origin.
Results
A New Classification Scheme of the Genetic Code
Figure 2 shows our new scheme for presenting the genetic code. It is based on a binary classification of nucleic acid bases. The two components of all nucleic acids, purines and pyrimidines, are denoted 1 and 0, respectively. The eight rows in Fig. 2 represent the 23 = 8 possible combinations of three binary digits. Since there are two purines (A, G) and two pyrimidines (U, C) for each row, there again exist eight possibilities.

A new classification scheme of the standard genetic code based on a binary representation of purines (1) and pyrimidines (0). The third base is given in parentheses. When there are differences between the standard code and any other code, the number of deviations from the standard code is indicated. This comparison is based on 16 nonstandard codes (Elzanowski and Ostell 2000). For instance, in the UG(G/A) field, 0/9 indicates that UGG encodes for Trp in all codes, but UGA is not the termination codon in 9 of the 16 nonstandard codes: In 8 different mitochondrial codes UGA encodes Trp, and in the euplotid nuclear code it represents Cys. It is interesting that at least in some bacteria the 21st amino acid, selenocysteine, can also be encoded by UGA (Osawa et al. 1992; Thanbichler and Böck 2002). Another example is the CU(G/A) field. In the yeast mitochondrion CUG and CUA encode Thr; in the alternative yeast nuclear code, CUG represents Ser. Shaded region show codon families. The point in the center indicates the perfect point symmetry in this scheme, according to Halitsky’s (2003) family–nonfamily symmetry operation. The thick horizontal line marks the symmetry axis for codon–anticodon symmetry.
Our first observation is that four (and not eight) columns are sufficient to place all 20 amino acids, as well as the termination codons. Each row contains exactly four different amino acids (including the termination codon). In the standard code, exceptions are the second row, with two leucines, and the AU* start codon in the fourth row. Note that here are also the deviations from the standard code. Interestingly, the yeast mitochondrial code shows no exception: Each row contains exactly four different entries in four different columns. In this respect the yeast mitochondrial code is the most regular one. The fact that in our scheme four columns are sufficient reflects the well-known fact that if the third position is important (in exactly half of our table this is not the case), then it is only decisive if there is either a purine (1) or a pyrimidine (0) (Fitch and Upper 1987), i.e., the third position is analyzed in a binary manner (Taylor and Coates 1989). This has been explained by Crick’s (1996) wobble hypothesis, wherein the first two nucleotides of the codon pair with their anticodon bases according to Watson–Crick rules, but the third base pairs according to the wobble rules, which say that G can also pair with U, for instance. The third codon position is exclusively analyzed in a binary manner in the mitochondrial codes of yeast, vertebrates, invertebrates, coelenterates, and flatworms, as well as in the codes of mold, protozoa, and mycoplasma/spiroplasma; for the other codes there are a few exceptions (cf. Elzanowski and Ostell 2000). Note that these few exceptions always have a purine at the third position of the codon (e.g., AUA [Ile] and AUG [Met] in the standard code).
Our scheme provides some support for the “adaptive genetic code” hypothesis (Freeland 2002), which states that the code has evolved to minimize the deleterious effects of mutation and translation error (Haig and Hurst 1991; Freeland and Hurst 1998). The purine–pyrimidine binary coding scheme, shown in Fig. 2, exhibits a much greater regularity than a binary coding according to the base pairs (A,U—1; G,C—0). This corresponds to the known fact that transition mutations (e.g., purine A vs. purine G) occur more frequently than transversion mutations (e.g., purine A vs. purimidine U).
A second observation concerns the order of the columns. In the first column the first two positions are G and C. These always pair with their anticodon base via three hydrogen bonds, i.e., the first two bases together always guarantee six hydrogen bonds. For that reason Lagerkvist (1978) called them strong codons. In the second and third columns, the first two bases guarantee five bonds (mixed codons), and in the fourth column just four bonds (weak codons). This pattern corresponds very well to the importance of the third base in the triplet codon: If the first bases are G and/or C (first column), the third base is never important, and in the second and third columns, the third base is important in exactly half the cases (if there is a purine in the second position—lower half of the table). In the fourth column the third base is always necessary for the determination of the correct amino acid. In Fig. 2, the order of codon families is illustrated by the shaded regions. It seems that for the first column, the first two bases alone guarantee sufficient stability in the codon–anticodon pairing to ensure the correct choice of the amino acid. In the case of mixed codons (second and third columns) a codon family is guaranteed if there is a pyrimidine in the second position. Going beyond Lagerkvist’s counting of hydrogen bonds, others have provided some quantitative information about nucleotide binding strengths (Ornstein and Fresco 1983).
A third observation refers to two perfect symmetries in our scheme. The first is the codon–anticodon symmetry: The thick horizontal line in Fig. 2 marks the symmetry axis. For instance, codon CCC (Pro; first column, first row) has the anticodon GGG (Gly; first column, last row), and codon ACG (Thr; third column, fourth row) has the anticodon UGC (Cys; third column, fifth row). The second perfect symmetry is the point symmetry corresponding to Halitsky’s (2003) family–nonfamily symmetry operation (“E–M bifurcation”), indicated by the point in the center of Fig. 2. Halitsky observed that all 32 “family codons” CC*, CU*, UC* GC*, GU*, AC*, CG*, and GG* can be mapped into the 32 “nonfamily codons” UU*, AU*, CA*, UG*, UA*, GA*, AG*, and AA* by exchanging the two amino bases A and C with one another and the two keto bases U and G with one another. For instance, the family codon GUA (Val) is mapped into the nonfamily codon UGC (Cys). Thus, this point symmetry underlies the family–nonfamily symmetry in our scheme (shaded vs. unshaded regions).
A fourth observation concerns the deviations of nonstandard genetic codes. As can be seen in Fig. 2, nearly all deviations occur in codons with a purine at the third position. The only exception is the yeast mitochondrial code, in which CU* does not code for Leu but, rather, for Thr.
Our fifth observation refers to the number of different tRNAs. The mammalian mitochondrial genomes contain one gene for each tRNA, with the exceptions of tRNA Leu and tRNA Ser for which two genes are present. Our new classification scheme for these mitochondrial codes (slight modification of Fig. 2) makes this number obviously: eight tRNAs for the eight codon families plus 14 tRNAs for the remaining 14 fields (the two termination codons need no tRNA).
Our sixth observation shows hitherto unknown regularities of amino acid properties. Jungck (1978) collected 15 different measures of amino acid properties, as well as 3 measures for dinucleoside monophosphates. For all of these 18 measures we arranged a table with eight rows and four columns corresponding to the scheme in Fig. 2. For AU(G/A) we took the Met values (e.g., vertebrate mitochondrial code); for UA(G/A), the Tyr values (mitochondrial flatworm code). Then we analyzed all row and column sums. The row sums show a strong monotonicity just for the three dinucleoside monophosphate measures and for the hydropohobicity measure of Levitt (1976). However, amazingly, the column sums of nearly all measures are perfectly correlated with the corresponding codon–anticodon binding strength as defined by Lagerkvist (1978, 1981), in the following simply denoted codon strength. This is demonstrated in Table 1. For this table we averaged the column sums of the second and third columns, giving one “mixed codons” column. As can be seen in Table 1 there are just two exceptions. In the polarity measure of Zimmerman et al. (1968), the deviation is very weak, and in contradiction to all other measures, here the values for the amino acids vary by orders of magnitude. A problem only arises for the three hydrophobicity measures: The two monotonic measures “Levitt” and “BullBreese” are anticorrelated, and the “Jones” measure is not monotonic. The anticorrelation was found by Jungck (1978), but he did not comment on this.
The fact that the order of the second and third columns is not fixed is also underlined by individual consideration of the two mixed codon columns, instead of the averaging done in Table 1. In about half of the cases the order of the second and third columns should be exchanged to guarantee the strong monotonicity of the amino acid measures as a function of the column number.
The strong correlation between amino acid properties and codon strength implies that the first and second position together, and not one of them alone, must have been important for the amino acid–codon assignment in the evolution of the genetic code.
Evolution of the Genetic Code
What do the observed patterns tell us about the evolution of the genetic code? The so-called biosynthetic theory assumes that the genetic code evolved from a simpler form that encoded fewer amino acids (Crick 1968). A special version of this theory has been given by Wong (1975), who proposes that the genetic code coevolved with the invention of biosynthetic pathways for new amino acids. Although it has been shown that his analyses rest on wrong assumptions (Ronneberg et al. 2000), it is generally accepted that one can discriminate evolutionarily old and new amino acids (Alberts et al. 2002). Of course it could be that the binding allocation between nucleic acid molecules (RNAs or even PNAs [Knight and Landweber 2000b]) and amino acids did not start until all 20 amino acids were available, but it seems simpler to assume that as soon as there were amino acids and nucleic acids available, produced abiotically, both began to bind to each other. It now seems clear that “the code probably underwent a process of expansion from relatively few amino acids to the modern complement of 20” (Knight and Landweber 2000b).
Does our scheme yield some hints as to the evolution of the code? We already noted that the third nucleotide is nearly always (two exceptions in the standard code) analyzed just in a binary manner. Taking this for granted, we can reduce our original 8 × 8 scheme to an 8 × 4 scheme (shown in Fig. 2). Looking at this scheme, we observe high redundancy for each second row. Therefore, it is tempting to speculate that there was a period during code evolution when the third position was not needed at all. Assuming this, we can cancel each second row and are left with a pure doublet code that encodes 4 × 4 = 16 amino acids (or 15 plus a termination codon). Perhaps, then, a doublet code preceded the triplet code, as has already been speculated (Jukes 1973; Hayes 1998).
Conceivably, codon expansion from doublet to triplet could have arisen before this or, possibly, not until all 16 amino acids were encoded. If one assumes the latter, then it is interesting to postulate for each doublet the corresponding old amino acid. Met (Wong 1975), Trp, Gln, Asn (Knight and Landweber 2000b), and Tyr (Alberts et al. 2002) seem to be newer amino acids. As mentioned above, Szathmary (1999) proposed an evolutionary mechanism of tRNA formation. In principle, this mechanism could also work starting with doublets instead of triplets. It should be possible to gain experimental evidence for a doublet code by studying amino acid–nucleic acid doublet binding in the same way as has been done for triplets. Knight and Landweber (2000a) showed that Arg triplet codons alone significantly associate with arginine binding sites. Perhaps the doublets show a higher specificity.
However, by proposing a doublet code one faces the frameshifting problem. It seems unthinkable that a sudden transition from a two-letter to a three-letter frame ever occurred. Instead, one can imagine a gradual evolution with an ancient three-letter reading frame where just the first two letters have been analyzed by an ancient translation machinery. However, one then wonders about such inefficient use of coding space. Perhaps the ancient translation machinery, simply for stereochemical reasons, could not analyze a two-letter frame. In this context it is also interesting to note that even our contemporary code is somehow “inefficient”: Already a quaternary doublet code can encode 16 amino acids (or 15 plus a termination codon). For just four (or five) further amino acids a third letter is necessary. Of course, this inefficiency has the advantage of robustness enhancing redundancy.
Szathmary (1992, 2003) proposed a model which yields the result that two different base pairs represent an optimal compromise between the overall copying fidelity and the overall reproduction rate (metabolic efficiency). He assumed that the genetic code was developed before evolution invented proofreading. For higher copying fidelity (due to proofreading, etc.), the model predicts that three different base pairs are better than just two. It is tempting to speculate that in the earliest phases of biological evolution with the lowest copying fidelity, just one base pair could have worked as well. (The copying fidelity is always highest for just one base pair. Nevertheless, Szathmary’s simple model gives no one-base pair optimum, but a more detailed model for the metabolic efficiency could do so.) So, perhaps, nucleic acid–amino acid mapping started with a binary code. This is in accordance with earlier speculations that the first genetic material contained only a single base-pairing unit (Crick 1968; Orgel 1968). An important argument in this context is the chemical instability of cytosine, so that it may be difficult to establish a genetic system with G–C base pairing (Levy and Miller 1998). Wächtershäuser (1988) proposed an all-purine precursor of nucleic acids. However, for the sake of self-replication it is more obvious to assume a two-letter code that can give rise to complementary base pairing. Jimenez-Sanchez (1995) argued for an early (binary) A–U coding. Recently, a ribozyme composed of only two different nucleotides has been found by in vitro evolution that contained the pyrimidine uracil and the purine 2,6-diaminopurine (Reader and Joyce 2002). Note that uracil is the biosynthetic precursor of the pyrimidines cytosine and thymine (the corresponding precursor of the purines adenine and guanine is hypoxanthine).
Of course, a binary encoding also would be the most aesthetic version from a purely mathematical point of view. A binary triplet code would represent just one column in our scheme (Fig. 2). Given the high redundancy between the rows, it is unlikely that this ever happened. However, an even simpler coding, a binary doublet code, seems conceivable. It is tempting to speculate which four amino acids, one per two consecutive rows, were the first encoded ones. In the first two rows (two pyrimidines, i.e., 00) Ser seems to be the oldest amino acid, and in the third and fourth rows (10), Ala (Wong 1975). On the other hand, the 01 rows obviously contain no really old amino acid, while the 11 rows contain more than one: Gly, Asp, and Glu (Wong 1975).
One could speculate that the termination marker was important from the very beginning and resulted in coding by the 01 binary doublet. It has been noted that the five amino acids coded by G** (Ala, Val, Gly, Asp, Glu) are all at or near the head of the amino acid synthesis pathways (Taylor and Coates 1989) and also the most abundantly formed ones in abiotic synthesis experiments (Miller 1953, 1987). Furthermore, it has been shown recently by extensive statistical analyses that the frequencies of all five G** amino acids are significantly higher in evolutionary conserved residues, and it has been concluded that “these amino acids may have been the first introduced into the genetic code” (Brooks and Fresco 2002, 2003; Brooks et al. 2002). This is also consistent with physicochemical arguments proposing that the first sense codons had the form G** (Eigen and Schuster 1978). However, Gly is biochemically built from Ser, so Ser can be assumed to be prior. It could be that in the beginning of nucleic acid–amino acid assignment, Asp and Glu competed for the 11 doublet. Of course, code transfer from one amino acid to another one might also have occurred (Wong 1975).
Another scenario consistent with a binary doublet code has been given by Fitch’s “ambiguity reduction” hypothesis (Fitch and Upper 1987). It states that early in evolution there was an ambiguity in the charging of amino acids to anticodon acceptors: In the first step just *pyrimidine* codons (*0*), coding for hydrophobic amino acids, and *purine* codons (*1*), coding for hydrophilic amino acids, have been distinguished (binary singulet code). In the second step the more refined binary doublet code (00*, 01*, 10*, 11*) evolved.
The idea that the doublet code was just the second state in the evolution of the genetic code, and that this evolution started with just the midbase as coding, has been worked out by others, who termed it the “simplet” code (McClendon 1986; Schwemmler 1994). However, in this hypothesis both old amino acids Ser (UC*) and Ala (GC*), as well as Asp (GA*) and Glu (GA*), cannot be discriminated. We therefore suggest that the first two positions were equally important from the very beginning. Although our suggestion also does not allow discrimination between the related amino acids Asp and Glu, it nevertheless allows discrimination between the functionally divergent amino acids Ser and Ala. A further argument for the evolutionary importance of the first two nucleotides is the strong correlation observed between codon strength and the amino acid properties.
Conclusion
Taylor and Coates (1989) stated that “many parts of the patterns (of the genetic code) have been seen by others but ... it is the synthesis that adds up to the most interesting ... new insights.” In this spirit, we note that in the work presented here different patterns appear more clearly than in the common scheme of the genetic code. An example is Lagerkvist’s (1978) observation that all strong codons represent codon families, while weak codons do not. Mixed codons represent codon families in half of the cases. Our presentation of the code also highlights new patterns, which were not seen before. As summarized in Table 1, nearly all measures of the amino acid properties correlate strongly with the codon strengths. Furthermore, there is perfect codon–anticodon symmetry as well as point symmetry corresponding to the family–nonfamily symmetry operation (Halitsky 2003) in our scheme.
With regard to evolution, we hypothesize that codon assignments started from a binary doublet code (e.g., hypoxanthin and uracil) and developed later to a quaternary doublet code (A, G, C, U); thereafter, expansion to a triplet code took place. Although the third position is needed for correct amino acid recognition, until now it has nearly always been analyzed in a binary manner. The conclusion that code evolution must have started with doublets and not with a single letter is also underlined by the correlation observed here between the properties of amino acids and the strengths of their codons.
References
S Alberti (1997) ArticleTitleThe origin of the genetic code and protein synthesis J Mol Evol 45 352–358
B Alberts A Johnson J Lewis M Raff K Roberts P Walter (2002) Molecular biology of the cel Garland Science New York
DH Ardell G Sella (2002) ArticleTitleNo accident: Genetic codes freeze in error-correcting patterns of the standard genetic code Phil Trans R Soc Lond B 357 1625–1642
I Barzilay JL Sussman Y Lapidot (1973) ArticleTitleFurther studies on the chromatographic behaviour of dinucleoside monophosphates J Chromatogr 79 139–146
DJ Brooks JR Fresco (2002) ArticleTitleIncreased frequency of cysteine, tyrosine, and phenylalanine residues since the last universal ancestor Mol Cell Prot 1 IssueID2 125–131
DJ Brooks JR Fresco (2003) ArticleTitleGreater GNN pattern bias in sequence elements encoding conserved residues of ancient proteins may be an indicator of amino acid composition of early proteins Gene 303 177–185
DJ Brooks JR Fresco AM Lesk M Singh (2002) ArticleTitleEvolution of amino acid frequencies in proteins over deep time: Inferred order of introduction of amino acids into the genetic code Mol Biol Evol 19 IssueID10 1645–1655
HB Bull K Breese (1974) ArticleTitleSurface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues Arch Biochem Biophys 161 665–670
FHC Crick (1966) ArticleTitleCodon-anticodon pairing: The wobble hypothesis J Mol Biol 19 548–555
FHC Crick (1968) ArticleTitleThe origin of the genetic code J Mol Biol 38 367–379
P Dunnill (1966) ArticleTitleTriplet nucleotide–amino acid pairing: A stereochemical basis for the division between protein and nonprotein amino acids Nature 210 1267–1268
M Eigen (1977) ArticleTitleThe hypercycle. A principle of natural self-organization. A: Emergence of the hypercycle Naturwissenschaften 64 541–565 Occurrence Handle1:CAS:528:DyaE1cXjtlGjtQ%3D%3D Occurrence Handle593400
M Eigen P Schuster (1978) ArticleTitleThe hypercycle: A principle of natural self-organization Naturwissenschaften 65 341–368
Elzanowski A, Ostell J (2000) Genetic codes. http://www3.ncbi.nlm.nih.gov/htbin-post/Taxonomy/wprintgc?mode = t#SG1
WM Fitch K Upper (1987) ArticleTitleThe phylogeny of tRNA sequences provides evidence for ambiguity reduction in the origin of the genetic code Cold Spring Harbor Symp Quant Biol 52 759–767 Occurrence Handle1:CAS:528:DyaL1cXlt1Cmtrc%3D Occurrence Handle3454288
SJ Freeland (2002) ArticleTitleThe Darwinian genetic code: An adaptation for adapting? Genet Progam Evolv Machines 3 113–127
SJ Freeland LD Hurst (1998) ArticleTitleThe genetic code is one in a million J Mol Evol 47 238–248 Occurrence Handle1:CAS:528:DyaK1cXmt1ansrs%3D Occurrence Handle9732450
SJ Freeland RD Knight LF Landweber LD Hurst (2000) ArticleTitleEarly fixation of an optimal genetic code Mol Biol Evol 17 511–518
JP Garel D Filliol P Mandel (1973) ArticleTitleCoefficients de partage d’aminoacides, 1978 nucleobases, nucleosides et nucleotides dans un systeme solvant salin J Chromatogr 78 381–391
R Grantham (1974) ArticleTitleAmino acid difference formula to help explain protein evolution Science 18S 862–864
D Haig LD Hurst (1991) ArticleTitleA quantitative measure of error minimization in the genetic code J Mol Evol 33 412–417 Occurrence Handle1:CAS:528:DyaK3MXmsleqtrs%3D Occurrence Handle1960738
Halitsky D (2003) Extending the (hexa-)rhombic dodecahedral model of the genetic code: The code’s 6-fold degeneracies and the orthogonal projections of the 5-cube as 3-cube. Contributed paper (983-92-151), American Mathematical Society, and personal communication
M Hasegawa T Miyata (1980) ArticleTitleOn the antisymmetry of the amino acid code table Orig Life 10 265–270
B Hayes (1998) ArticleTitleThe invention of the genetic code Am Sci 86 8–14
JJ Hopfield (1978) ArticleTitleOrigin of the genetic code: A testable hypothesis based on tRNA structure, sequence, and kinetic proofreading Proc Natl Acad Sci USA 75 4334–4338
A Jimenez-Sanchez (1995) ArticleTitleOn the origin and evolution of the genetic code J Mol Evol 41 712–716
DD Jones (1975) ArticleTitleAmino acid properties and side-chain orientation in proteins: A cross correlation approach J Theor Biol 50 167–183
TH Jukes (1973) ArticleTitlePossibilities for the evolution of the genetic code from a preceding form Nature 246 22–26
JR Jungck (1971) ArticleTitlePre-Darwinian and non-Darwinian evolution of proteins Curr Mod Biol 3 307–318
JR Jungck (1978) ArticleTitleThe genetic code as a periodic table J Mol Evol 11 211–224
RD Knight LF Landweber (1998) ArticleTitleRhyme or reason: RNA-arginine interactions and the genetic code Chem Biol 5 R215–R220
RD Knight LF Landweber (2000a) ArticleTitleGuilt by association: The arginine case revisited RNA 6 499–510
RD Knight LF Landweber (2000b) ArticleTitleThe early evolution of the genetic code Cell 101 569–572
RD Knight SJ Freeland LF Landweber (2001) ArticleTitleRewiring the keyboard: Evolvability of the genetic code Nat Rev Genet 2 49–58
U Lagerkvist (1978) ArticleTitle“Two out of three”: An alternative method for codon reading Proc Natl Acad Sci USA 75 1759–1762
U Lagerkvist (1981) ArticleTitleUnorthodox codon reading and the evolution of the genetic code Cell 23 305–306
M Levitt (1976) ArticleTitleA simplified representation of protein conformations for rapid simulation of protein folding J Mol Biol 104 59–107
M Levy SL Miller (1998) ArticleTitleThe stability of the RNA bases: Implications for the origin of life Proc Natl Acad Sci USA 95 7933–7938
N Maizels AM Weiner (1987) ArticleTitlePeptide-specific ribosomes, genomic tags, and the origin of the genetic code Cold Spring Harb Symp Quant Biol 52 743–749
JH McClendon (1986) ArticleTitleThe relationship between the origins of the biosynthetic paths to the amino acids and their coding Orig Life 16 269–270
TL McMeekin ML Groves NJ Hipp (1964) Refractive indices of amino acids, proteins and related substances JA Stekol (Eds) Amino acids and serum proteins American Chemical Society Washington, DC 54–66
SL Miller (1953) ArticleTitleProduction of amino acids under possible primitive earth conditions Science 117 528–529
SL Miller (1987) ArticleTitleWhich organic compounds could have occurred on the prebiotic earth? Cold Spring Harb Symp Quant Biol 52 17–27
LE Orgel (1968) ArticleTitleEvolution of the genetic apparatus J Mol Biol 38 381–393
RL Ornstein JR Fresco (1983) ArticleTitleCorrelation of Tm, sequence, and ΔH of complementary RNA helices and comparison with DNA helices Biopolymers 22 2001–2016
S Osawa TH Jukes K Watanabe A Muto (1992) ArticleTitleRecent evidence for the evolution of the genetic code Microbiol Rev 56 IssueID1 22–264
JS Reader GF Joyce (2002) ArticleTitleA ribozyme composed of only two different nucleotides Nature 420 841–844
TA Ronneberg LF Landweber SJ Freeland (2000) ArticleTitleTesting a biosynthetic theory of the genetic code: Fact or artifact? Proc Natl Acad Sci USA 97 13690–13695
W Schwemmler (1994) Reconstruction of cell evolution: A periodic system of cells CRC Press Boca Raton, FL
TM Sonneborn (1965) Degeneracy of the genetic code: extent, nature, and genetic implications V Bryson HJ Vogel (Eds) Evolving genes and proteins Academic Press New York 297–377
E Szathmary (1992) ArticleTitleWhat is the optimum size for the genetic alphabet? Proc Natl Acad Sct USA 89 2614–2618
E Szathmary (1993) ArticleTitleCoding coenzyme handles: A hypothesis for the origin of the genetic code Proc Natl Acad Sci USA 90 9916–9920
E Szathmary (1999) ArticleTitleThe origin of the genetic code Trends Genet 15 223–229
E Szathmary (2003) ArticleTitleWhy are there four letters in the genetic alphabet? Nat Rev Genet 4 995–1001
FJR Taylor D Coates (1989) ArticleTitleThe code within the codons BioSystems 22 177–187
M Thanbichler A Böck (2002) ArticleTitleThe function of SECIS RNA in translational control of gene expression in Escherichia coli EMBO J 21 6925–6934
MD Topal JR Fresco (1976) ArticleTitleBase pairing and fidelity in codon-anticodon interaction Nature 263 289–293
G Wächtershäuser (1988) ArticleTitleAn all-purine precursor of nucleic acids Proc Natl Acad Sci USA 85 1134–1135
AL Weber JC Lacey SuffixJr (1978) ArticleTitleGenetic code correlations: amino acids and their anticodon nucleotides J Mol Evol 17 273–284
AM Weiner N Maizels (1987) ArticleTitletRNA-like structures tag the 3′ ends of genomic RNA molecules for replication: Implications for the origin of protein synthesis Proc Natl Acad Sci USA 84 7383–7390
CR Woese (1965) ArticleTitleOn the evolution of the genetic code Proc Natl Acad Sci USA 54 1546–1552 Occurrence Handle1:CAS:528:DyaF28Xltlagsg%3D%3D Occurrence Handle5218910
CR Woese (1967) The genetic code: The molecular basis for genetic expression Harper and Row New York
CR Woese DH Dugre WC Saxinger SA Dugre (1966) ArticleTitleThe molecular basis for the genetic code Proc Natl Acad Sci USA 55 966–974
CR Woese DH Dugre SA Dugre M Kondo WC Saxinger (1967) ArticleTitleOn the fundamental nature and evolution of the genetic code Cold Spring Harbor Symp Quant Biol 31 723–736
RV Wolfenden PM Cullis CCF Southgate (1979) ArticleTitleWater, protein folding, and the genetic code Science 206 575–577
JT-F Wong (1975) ArticleTitleA co-evolution theory of the genetic code Proc Natl Acad Sci USA 72 1909–1912 Occurrence Handle1:CAS:528:DyaE2MXkt1ertbg%3D Occurrence Handle1057181
M Yarus (1998) ArticleTitleAmino acids as RNA ligands: A direct-RNA-template theory for the code’s origin J Mol Evol 47 109–117
M Yarus (2000) ArticleTitleRNA-ligand chemistry: A testable source for the genetic code RNA 6 475–484
E Zuckerkandl L Pauling (1965) Evolutionary divergence and convergence in proteins V Bryson HS Vogel (Eds) Evolving genes and proteins Academic Press New York 97–167
JM Zimmerman N Eliiezer R Simna (1968) ArticleTitle. J Theor Biol 21 170–201
Acknowledgments
We thank two anonymous reviewers for many valuable comments and for referring us to relevant literature and A. Beyer, F. Grosse, M. Friedel, and M.-L. Merten for critical reading of the manuscript. This work was supported by Grant 0312704E from the Bundesministerium für Bildung und Forschung.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wilhelm, T., Nikolajewa, S. A New Classification Scheme of the Genetic Code. J Mol Evol 59, 598–605 (2004). https://doi.org/10.1007/s00239-004-2650-7
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2650-7