
Abstract
Thermodynamic stability and mutational robustness of secondary structure are critical to the function and evolutionary longevity of RNA molecules. We hypothesize that natural and artificial selection for functional molecules favors the formation of structures that are stable to both thermal and mutational perturbation. There is little direct evidence, however, that functional RNA molecules have been selected for their stability. Here we use thermodynamic secondary structure prediction algorithms to compare the thermal and mutational robustness of over 1000 naturally and artificially evolved molecules. Although we find evidence for the evolution of both types of stability in both sets of molecules, the naturally evolved functional RNA molecules were significantly more stable than those selected in vitro, and artificially evolved catalysts (ribozymes) were more stable than artificially evolved binding species (aptamers). The thermostability of RNA molecules bred in the laboratory is probably not constrained by a lack of suitable variation in the sequence pool but, rather, by intrinsic biases in the selection process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the short term, natural selection favors individual organisms that are best able to survive and reproduce. In the long term, natural selection favors lineages with consistently high fitness. What does it take for a lineage to be consistently fit? First, organisms must be able to withstand environmental perturbations either by remaining unperturbed by those changes or through adaptive genetic or phenotypic modifications (Meyers and Bull 2002). Second, lineages must be somewhat robust to the effects of random mutation.
This is true even at the level of a single biopolymer. Naturally occurring nucleic acids and proteins have, for the most part, evolved to fold, bind, and catalyze reactions robustly, regardless of the environment in which organisms find themselves. In addition, the nucleic acid replication machinery allows for occasional variation (or error), which is essential for producing the novelty that fuels evolution. Much of the time, however, such mutations have deleterious effects on the form and function of a molecule. Biopolymers that are highly sensitive to mutations may be unreliable, and are eventually eliminated by natural selection (van Nimwegen et al. 1999; Wagner et al. 1997).
There is evidence that these two forms of stability—structural/functional stability and mutational stability—are biophysically correlated for both RNA and proteins, and that the evolution of one form of robustness goes hand-in-hand with the evolution of the other form (Ancel and Fontana 2000; Bornberg-Bauer and Chan 1999; Bussemaker et al. 1997; Vendruscolo et al. 1997). The term plastogenetic congruence was coined to describe a more general correlation between the effects of the environment and the effects of mutation on form and function (Ancel and Fontana 2000). In RNA, plastogenetic congruence means that the set of shapes that are thermodynamically accessible to a molecule overlaps significantly with the set of groundstate shapes produced by point mutations to that molecule. In particular, if one or more alternative shapes (other than the groundstate) have thermodynamic stabilities similar to the groundstate, then there will likely also be one or more single mutations that will stabilize these alternative shapes relative to the groundstate. If, instead, a molecule folds stably into its groundstate, in the absence of any comparably stable suboptimal shapes, then most mutations to the molecule will leave that structure unchanged. Conversely, the diversity of groundstates produced by point mutations to a molecule is indicative of the diversity of shapes thermodynamically accessible to the original molecule. This biophysical link between thermodynamic and genetic stability implies that the evolution of thermodynamic robustness necessarily entails genetic robustness and vice versa (Ancel and Fontana 2000).
Sequences that form stable structures are likely to be more functional than sequences that can flit between various structures. Thus we suspect that any biopolymer under natural or artificial selection to perform a function should show some degree of thermostability. Nonetheless, naturally evolved biopolymers that are the products of eons of exposure to the vagaries of the environment and error-prone replication should show greater thermostability than biopolymers that have been evolved in vitro and have thus been selected in a much more controlled environment over a much shorter period of time. By virtue of both plastogenetic congruence and years of natural selection against deleterious mutations, we similarly expect greater mutational robustness in natural molecules than in those selected in the lab. In order to test the hypothesis that naturally evolved molecules should be much more mutationally and thermodynamically robust than the products of directed selection, we use computational algorithms to estimate the thermodynamic stability and mutational stability of a series of natural, functional RNA molecules, including tRNAs, 5S rRNAs, and natural ribozymes, and compare these to the stabilities of a large number of artificially selected nucleic acid binding species (aptamers) and catalysts (ribozymes) that have been collected in an Aptamer Database, now available online.
Materials and Methods
The Sequences
The current version of the Aptamer Database contains entries from 237 papers that describe the in vitro selection of aptamers, ribozymes, and deoxyribozymes. The database primarily contains catalysts that have been selected from completely random pools, as opposed to variants on known, natural ribozymes. Each entry is described by the following fields: Author (last name and first name of the authors of each publication); Title; Medline Accession Number (allowing a direct link to the Pubmed record); Target (name of the ligand that was used for selection); Target Type (currently classified into proteins, peptides, nucleic acids, organic molecules, inorganic molecules or other); Journal (year, volume, issue, pages); DNA/RNA or Modified (indicates whether the initial nucleic acid pool used for the selection was ‘natural’ or contained modified nucleotides); Buffer Conditions; Template Description (describes the length of the random region); Template Sequence (describes the sequences of the terminal primer binding sites); Sequences (list of each aptamer or ribozyme sequence that was selected, not including the Template Sequence or constant region).
The database is updated monthly. Data are entered manually into the database. Each sequence is entered twice and the two entries are compared to ensure accuracy. The Aptamer Database is publicly available through aptamer.icmb.utexas.edu. To facilitate data entry by other users, a template of the database is available for download at aptamer.icmb.utexas/submit.
We computed the thermodynamic and mutational stability of 532 aptamers and 209 ribozymes from aptamer.icmb.utexas.edu; 193 bacterial tRNAs, 79 nonhuman eukaryotic tRNAs, and 73 human tRNAs from rna.wustl.edu/GtRDB/ (Lowe and Eddy 1997); 47 archaebacterial 5S rRNAs, 67 eubacterial 5S rRNAs, and 168 eukaryotic 5S rRNAs from biobases.ibch.poznan.pl/5SData/ (Szymanski et al. 2000); and 304 hammerhead ribozyme sequences from www.sanger.ac.uk/cgi-bin/Rfam/getacc?RF00008 (Griffiths-Jones et al. 2003). For each of these molecules, we generated 1000 random molecules, preserving base composition and length. In the case of the in vitro selected aptamers and ribozymes, we randomized only the regions that were originally, experimentally randomized, whereas for the naturally occurring molecules, we randomized the entire sequence.
Measuring Thermodynamic Stability
We computationally estimated the extent to which single molecules are buffered against thermodynamic noise. First, we computed the repertoire of structures that are accessible to a sequence and approximated the probability of each structure. We used an extension of the standard thermodynamic minimum free energy folding algorithm, which permits the computation of all secondary structures within some energy range above the minimum free energy (Hofacker et al. 1994; Wuchty et al. 1999). This algorithm provides reasonable approximations but is by no means perfect. It does not predict pseudoknots or other tertiary interactions, which are known to occur in both natural and artificial RNA molecules. The thermodynamic parameters used by the algorithm are taken from( Mathews et al. 1999; Walter et al. 1994) and have been estimated to predict the correct structure 73% of the time (Mathews et al. 1999; Walter et al. 1994). The parameters are estimated at the physiological temperature of 37°C, which is appropriate for prediction of natural sequences. Artificial RNAs, however, are often selected and optimized at a lower temperature (25°C). These algorithms may thus make less accurate predictions for artificially selected molecules.
Despite these limitations, we used the suboptimal folding algorithm to rapidly approximate the low energy portion of the secondary structure space of a given sequence. We neglected energy barriers and assumed that a sequence equilibrates among all structures whose free energy is within 5 kT of the groundstate. The 5-kT choice amounts to approximately 3 kcal at 37°C and corresponds to the loss of two CG/GC stacking interactions.
Under thermodynamic equilibration, we assumed that the Boltzmann probability of a shape s, exp(−ΔG σ/kT)/Z, approximates the overall fraction of time that the molecule spends in s, where ΔG σ is the free energy of structure s, k is the Boltzmann constant, T the absolute temperature, and Z = ∑τ exp(−ΔG τ/kT) is the partition function, which is computed by an algorithm described by McCaskill (1990). We used the Boltzmann probability of the lowest free energy state (the groundstate) to estimate the thermodynamic stability of a molecule. Thus we did not use minimum free energy alone to estimate thermostability but, instead, considered the extent to which a groundstate is stabilized with respect to competing shapes.
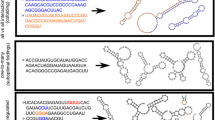
In measuring thermodynamic stability, we assayed secondary structures at two levels of resolution. As illustrated in Fig. 1, the standard secondary structure of a molecule contains the pairing status of every individual base. The coarse grained secondary structure of a molecule notes the relative position of the following structural components: hairpins (H), interior loops (I), bulges (B), multiloops (M), and stacks (S). We first predicted the standard structure of a molecule, and then parsed the standard representation into these five components. When several suboptimal standard shapes reduced the same coarse-grained shape, we grouped them together and summed their Boltzmann coefficients. The coarse-grained groundstate was the shape with the largest (collective) Boltzmann, which may or may not have corresponded with the original standard groundstate.

Two levels of structural resolution. Standard secondary structure (left) contains the pairing status of every nucleotide, whereas coarse-grained structure (right) indicates the relative positions of structural components including hairpins (H), stacks (S), and multiloops (M).
In essence, the standard secondary structure looks at the precise sequence and structure of a molecule, while the coarse-grained structure is a measure of the overall fold, independent of sequence. By examining coarse-grained structures, we grouped structurally similar molecules. This may be appropriate, for example, if two nucleic acids are functionally similar because of a shared stacked helical junction, but these junctions in the two molecules differ in the number of single-stranded residues separating the two participating helices. Such molecules would be substantially different in the standard representation but appropriately equivalent in the coarse-grained representation. Coarse-grained structures also allow for some degree of error in the thermodynamic predictions and, thereby, provide an additional check on the validity of the analysis.
Measuring Mutational Robustness
For every molecule, we measured mutational neutrality, which is the fraction of all possible point mutations that perturb the groundstate. A molecule of length L will have 3L one-error mutants (sequences that differ by exactly one mutation). We folded the (standard and coarse-grained) groundstate of each of these mutants and tallied those that preserve the original groundstate. This indicated the extent of intrinsic mutational buffering. We did not consider structural changes that require multiple simultaneous mutations.
Measuring Plastogenetic Congruence
We estimated the relationship between thermostability and mutational robustness by calculating the correlation coefficient for thermostability and mutational robustness. We report r and its 95% confidence intervals.
Measuring Thermostability Potential
In order to assess whether artificially selected molecules might evolve further stability given time, we calculated the groundstate shape and thermodynamic stability of every one-error mutant. We report the fraction of one-error neighbors that either preserve or further stabilize the original groundstate shape and the extent to which stability is increased by these mutations. We are interested in whether laboratory selection fails to select more thermostable variants or whether such variants do not exist in the first place.
We report the likelihood that a single point mutation will further stabilize the existing groundstate. A high value suggests that more stable mutants are likely to exist in a random library and arise under mutagenesis and that the failure to select such variants is perhaps due to a lack of selection pressure for thermostability or even selection against thermostability.
Results and Discussion
Thermostability and Mutational Robustness ofRandom Sequences
The length and composition of a given sequence will likely affect its thermodynamic stability and mutational robustness. As proof of this, we computationally generated 1000 different random sequences for multiple different lengths and base compositions and then measured and plotted the (standard structural) thermodynamic stability and mutational robustness for each sequence (Fig. 2). As previously noted, there is an obvious and general correspondence between thermodynamic stability and mutational robustness. While sequences of all lengths and composition demonstrated this trend, however, the degree of thermodynamic stability and mutational robustness varied as a function of sequence length and composition.

Sequence length, base composition, and stability. Thermostability is estimated with the Boltzmann coefficient of the groundstate (top); mutational robustness is the fraction of point mutations that leave the groundstate unchanged (middle); and platogenetic congruence is the correlation between these two forms of stability (bottom). The top two graphs in each panel show average and 95% confidence intervals and the bottom graph shows the correlation coefficient for thermostability and mutational robustness (r) and 95% confidence intervals based on 1000 random sequences of (A) each length (40, 80, and 120 bases) and (B) each base composition (20, 30, 40, 50, and 60% GC). Inset: Stability for a set of 1000 sequences of (A) length 40 and (B) 70% GC.
Previously, Ancel and Fontana (2000) have suggested that a correlation exists between thermostability and mutational stability, and have termed this correlation plastogenetic congruence. In other words, the more stable a structure is to denaturation by heat, the more stable it may be to disruption by mutation. The correlation between thermostability and mutational robustness is statistically significant (p < 0.0005) for all sequence lengths, although the extent of thermostability and mutational robustness concurrently decrease with length (Fig. 2A). The reduced ability of longer sequences to readily maintain a given structure (and hence a given function) in response to either thermal or mutational pressure may have been a boon in the evolution of new structures and functions early in evolution (James and Ellington 1999; Levy and Ellington 2001; Schultes and Bartel 2000). The relationship between thermostability and mutational stability is more complex when examined as a function of GC content (Fig. 2B). As might be expected, thermostability increases with GC content. Mutational stability, however, slightly decreases. Nonetheless, within each GC content class, there is significant plastogenetic congruence, that is, a significant positive correlation between thermostability and mutational stability (p < 0.0001). Interestingly, these results overall suggest that an organism in the putative “RNA world” (Gilbert 1986) whose metabolism relied predominantly upon functional nucleic acids might have “tuned” its evolvability (sensitivity to mutation) by tuning its GC content.
Thermostability and Mutational Robustness ofEvolved Sequences
In order to normalize for length and compositional biases that might arise from the diverse, real data sets under consideration, we generated sets of 1000 random molecules with identical length and composition for each naturally and artificially selected molecule. We refer to these molecules as random (1000 scrambled variants) and original (single, parental variant), respectively. Using standard thermodynamic prediction algorithms from the Vienna package (Hofacker et al. 1994), every original and randomized molecule is analyzed for its standard and coarse-grained thermodynamic stability and standard and coarse-grained mutational stability.
The original molecule will be more stable than some subset of its 1000 random counterparts and less stable than the rest. For all four measures of stability, we calculate the percent of random sequences that are less stable than the true sequence. This statistic—the percentile of the original molecule—indicates whether the original molecule is more stable than expected while normalizing for both length and base composition. Any value above 0.5 indicates that a molecule is more stable than would be expected by chance alone. This is a conservative normalization. Although base composition could presumably evolve in response to selection for thermostability, it may be significantly constrained by other factors such as competition for metabolic resources, ultraviolet exposure, replication error, and repair efficiency (Calcagnile et al. 1996; Cox and Yanofsky 1967; Rocha and Danchin 2002). Such constraints may lead to differences in thermostability among taxa that are not actually the result of differential selection for thermostability. Note that these calculations differ substantially from those shown in Fig. 2, as do the axes in the attendant graphs.
In general, both natural and artificially selected molecules are more stable than would have been expected by chance, and the results from standard and coarse-grained stability analyses are largely in agreement with one another (Fig. 3A). Surprisingly, different types of functional RNA molecules from different classes of organisms show a wide range of thermostabilities. Nonetheless, it is generally true that natural, functional RNA molecules are more thermostable than artificially selected molecules such as aptamers and ribozymes. This correlation is true even when other factors might be expected to interfere. For example, all types of tRNAs show greater thermostabilities than artificially selected molecules. More so than almost any other functional RNAs, the stabilities of tRNA molecules are due to tertiary structural interactions, and posttranscriptional modifications frequently contribute to stabilization. For example, Wutchy et al. (1999) showed that modification significantly increased the predicted thermostability of an E. coli tRNAlys molecule. We nevertheless analyze the unmodified tRNA sequences, as they are functional prior to modification (Claesson et al. 1990; Harrington et al. 1993; Pestova and Hellen 2001; Sampson and Uhlenbeck 198S; Takai et al. 1996) and make for an interesting intermediate between artificially selected molecules that have undergone only brief selection for thermostability and naturally evolved molecules that are not modified or significantly stabilized by external forces. One might expect that the secondary structures of tRNAs would be under less selective pressure than the secondary structures of aptamers and ribozymes, since the functions of these artificially selected molecules derive largely from unmodified stem loops or other common secondary structural elements. Figure 3B illustrates an overall difference between sequences evolved in the lab and sequences evolved in nature and confirms that artificially selected sequences are less stable, under both standard and coarse-grained comparisons.

Comparative thermostability of RNA. A Average percentile thermostability for nine classes of RNA. Apt—artificial aptamers; Ribo—artificial ribozymes; Eub tRNA—eubacterial tRNA; Euk tRNA—nonhuman eukaryotic tRNA; Hum tRNA—human tRNA; Arch 5S—archaebacterial 5S rRNA; Eub 5S—eubacterial 5S rRNA; Euk 5S—eukaryotic 5S rRNA; Hammer—natural hammerhead ribozymes. We use the Boltzman coefficient of the minimum free energy shape to estimate thermostability. The percentile is the rank of a sequence’s thermostability compared to the thermostabilities of 1000 randomly generated sequences of the same length and base composition. Percentiles significantly above 0.5 are more thermostable than expected from a random sequence. Standard and coarse grained refer to the level of structural resolution as depicted in Fig. 1. Bars indicate 95% confidence intervals. B Percentile thermostabilities are averaged over all artificially evolved aptamers and ribozymes and over all seven classes of natural tRNA, rRNA, and ribozymes.
While this discrepancy is the most clear and intuitive result of our analysis, we also venture to speculate on the observed variation among different types of RNA molecules. For example, artificially selected ribozymes are significantly more stable than artificially selected aptamers. This may represent the greater functional (and therefore possibly structural) constraints on catalysts relative to binding species. It is more difficult to frame a direct comparison between hammerhead ribozymes and other natural, functional RNA molecules, since ribosomal and transfer RNAs perform a variety of functions other than just binding to proteins. Other differences are more enigmatic: eubacterial 5S rRNA molecules have surprisingly low stabilities relative to the higher stabilities of eukaryotic and archaebacterial 5S rRNAs. Hammerhead ribozyme sequences show somewhat higher stabilities than artificially evolved ribozymes under standard folding, but their relative stabilities are lower under coarse-grained folding.
There is a less pronounced relationship between the evolutionary or functional history of a molecule and its mutational stability. Figure 4A shows artificially selected aptamers and ribozymes to be statistically indistinguishable from each other. The naturally selected sequences differ significantly in mutational rigidity from the artificially selected sequences, in both directions. Notably human tRNAs show much greater mutational robustness than artificially selected sequences, while nonhuman eukaryotic tRNAs as a whole are less mutationally robust. As was the case with computationally generated sequences (Fig. 2), while there is a general correlation between thermostability and mutational stability (plastogenetic congruence), the correlation is not exact; for example, eukaryotic tRNA molecules are relatively thermostable but not similarly stable to mutation. Nonetheless, when the data sets are combined there is again an overall, statistically significant discrepancy between naturally and artificially selected molecules, with naturally selected RNAs showing a greater propensity to retain their secondary structures when challenged with mutations (Fig. 4B).

A Comparative mutational robustness of RNA. Average neutrality percentile for nine classes of RNA. (See legend to Fig. 3.) We use neutrality—the fraction of single-point mutations that preserve the minimum free energy structure—to estimate mutational robustness. The percentile is the rank of a sequence’s neutrality compared to the neutralities of 1000 randomly generated sequences of the same length and base composition. Percentiles significantly above 0.5 are more mutationally robust than expected from a random sequence. Standard and coarse grained refer to the level of structural resolution as depicted in Fig. 1. Bars indicate 95% confidence intervals. B Robustness percentiles are averaged over all artificially evolved aptamers and ribozymes and over all seven classes of natural tRNA, rRNA, and ribozymes.
Finally, using a subset of 100 aptamers and 100 archaebacterial 5S rRNA molecules, we calculated the fraction of point mutations that would further stabilize the original structure. Archaebacterial 5S rRNA molecules were chosen as the comparison set since they were initially found to be significantly more thermostable and mutationally stable than aptamers. The average fraction of structurally beneficial mutations was not statistically different for the aptamers (0.123812) and archaebacterial 5S rRNAs (0.132992). However, the degree to which structurally beneficial mutations further stabilized the original structure was significantly divergent (Fig. 5). As was predicted based on their lower thermostabilities and mutational stabilities, aptamers showed significantly more capacity for additional stabilization than did 5S archaebacterial RNAs.

Potential increase in thermostability due to point mutation. Max improvement is the maximum improvement in thermostability achieved by a point mutation, measured as a multiple of the original stability. Avg improvement is the average increase in thermostability across all mutations that maintain and further stabilize the orginal groundstate.
Overall, these results suggest that artificially selected molecules are less thermodynamically and mutationally robust than naturally occurring molecules. Intuitively, artificially selected molecules have experienced a much more consistent environment over much shorter periods of time than have natural molecules. In consequence, thermostability and especially mutational robustness may have been mostly irrelevant for the function for which they were selected. In fact, thermostability may even be costly in vitro. Selection experiments rely upon the polymerase chain reaction for the amplification of functional species, and excessive structural stability may lead to discrimination between amplicons. This limitation has likely already been seen in a selection for oligonucleotide substrates for T4 DNA ligase from a random sequence population (Harada and Orgel 1993). Despite the well-known propensity of ligase to covalently join substrates that are paired with a DNA template, these experiments yielded substrates that were mismatched with the template, potentially because perfectly paired substrates were at a selective disadvantage during amplification. Similarly, in vitro selection of an RNA ligase produced molecules that formed alternative, inactive conformations, possibly because such conformational flexibility facilitated replication. Upon subsequent reselection under more stringent selection conditions, the ligase evolved greater structural stability and a lower propensity for misfolding (Schmitt and Lehman 1999).
In contrast, structural stability may be under intense selection in natural, functional RNA molecules. For example, a comparison among ribosomal RNAs of related free-living and endosymbiotic bacteria indicated that the endosymbiont RNAs contain mutations that generally lead to structural destabilization (Lambert and Moran 1998). Such destabilizing mutations have not persisted in free-living populations, presumably because the fitness costs are more severe. Similarly, endosymbiont tRNAs are by and large less structurally stable than the tRNAs of theirfree-living counterparts (Lynch 1996, 1997). RNA processing mechanisms in natural systems may therefore have evolved to promote thermostability.
Just as there are examples of naturally evolved thermostability, there are also naturally occurring RNA molecules with structural flexibility. The Hepatitis Delta Virus (HDV) ribozyme is thought to assume several different conformations, some active and some inactive (Gottlieb et al. 1994; Smith et al. 1992). Similarly, slow in vivo transcription rates allow the potato spindle tuber viroid to form sequential, metastable structures (Repsilber et al. 1999). Most recently, a number of RNA regulatory elements (so-called “riboswitches”) have been shown to undergo conformational changes in the presence of metabolites (Lai 2003). Our tools for assessing the thermostability and mutational robustness may ultimately shed light on the molecular underpinnings of functional RNAs with metastable structures or alternative conformations.
Practical Implications
While directed evolution is a powerful tool, it apparently does not optimize nucleic acid structures to the same extent as billions of years of natural selection. Based on the analyses summarized in Fig. 5, there appeared to be numerous aptamer variants that would have retained a functional structure and been more thermodynamically stable. It is possible that these more stable variants did not predominate in the selection experiments because the mutant sequences were not originally present in the population or did not arise during the course of amplification. However, given that many nucleic acid pools are thought to initially have relatively complete coverage of sequence spaces up to 25 nucleotides in length, the lack of more stable variants may instead be indicative of a general discrimination against the most stable structures during amplification.
In order to further examine this hypothesis, we examined the thermodynamic and mutational stabilities of a series of RNA catalysts that were selected from a random sequence pool for their ability to cleave a RNA substrate (Fig. 6) (Salehi-Ashtiani and Szostak 2001). This experiment was especially informative because the sequences and catalytic abilities of the self-cleaving ribozymes were determined at several points during the selection. Ultimately, the selection experiment yielded RNA catalysts whose sequences and structures were by and large similar to that of the hammerhead ribozyme. We therefore can directly compare the thermostabilities and mutational stabilities of the artificially selected RNAs to those of natural hammerhead ribozymes. As can be seen in Fig. 6, the early rounds of selection generated artificial catalysts with thermostabilities greater than in the starting pool and approximately equal to that of natural hammerhead ribozymes. As the selection progressed, however, the thermal and mutational stabilities of the artificially selected ribozymes actually decreased, even though the overall activity of the population progressively increased. While these results might indicate that functional superiority is inversely correlated with structural stability, such an insight would be extremely counterintuitive. It seems more likely that there were many highly functional catalysts in the stable Round 5 population but that only those catalysts that could also meet the requirements of replicability survived through Round 12. That is, to the extent that thermal stability of a RNA molecule makes it less replicable, then functional but less replicable molecules may ultimately be chosen in a selection experiment.

Thermostability during in vitro evolution of hammerhead ribozymes. The average thermostability percentile (see legend to Fig. 3) for sequences taken from consecutive rounds of in vitro selection. The selected molecules begin unstable, hit peak stability in round 9, and then lose stability in later rounds of selection. Round 9 thermostability is comparable to that found in natural hammerhead ribozymes. The mutational robustness of the selected populations follows a qualitatively similar trajectory.
This analysis suggests some extremely practical considerations for biopolymer engineers. First and most importantly, including thermostability as a criterion during the directed evolution of nucleic acids may result in unexpected improvements in functionality. This hypothesis can now be directly evaluated by carrying out parallel selection experiments at different temperatures. Second, the pool sizes used for selection experiments may be artificially constrained by the amplification process, a finding that would skew the interpretation of many published results that speculate on the relative difficulties of identifying functional nucleic acid molecules during the origin and evolution of metabolism. Third, many molecules evolved in the lab may eventually be tasked in natural environments. While predicting all possible natural challenges for an engineered molecule would be next to impossible, one could easily modify selection protocols to favor thermostability and, thereby, hedge against a diversity of environmental insults. For example, Bevilacqua and co-workers have carried out a number of selections for hairpin structures with increased thermostabilities (Bevilacqua and Bevilacqua 1998; Moody and Bevilacqua 2003; Nakano et al. 2002; Proctor et al. 2002; Shu and Bevilacqua 1999). Interestingly, a number of the hairpin loops that were selected for thermostability resemble those that also occur in natural RNA structures, including RNA molecules from mesophiles. Similarly, Guo and Cech selected for increased thermostability in variants of the Tetrahymena Group I self-splicing ribozyme (Guo and Cech 2002). In this case, the mutations were predominantly in tertiary structural contacts rather than secondary structures, as might be expected for a large and complex ribozyme structure.
References
LW Ancel W Fontana (2000) ArticleTitlePlasticity, evolvability and modularity in RNA J Exp Zool (Mol Dev Evol) 288 242–283 Occurrence Handle10.1002/1097-010X(20001015)288:3<242::AID-JEZ5>3.0.CO;2-O Occurrence Handle1:CAS:528:DC%2BD3cXnsFChs70%3D
JM Bevilacqua PC Bevilacqua (1998) ArticleTitleThermodynamic analysis of an RNA combinatorial library contained in a short hairpin Biochemistry 37 15877–15884 Occurrence Handle10.1021/bi981732v Occurrence Handle1:CAS:528:DyaK1cXmsleitbs%3D Occurrence Handle9843393
E Bornberg-Bauer HS Chan (1999) ArticleTitleModeling evolutionary landscapes: Mutational stability, topology, and superfunnels in sequence space Proc Natl Acad Sci USA 96 10689–10694 Occurrence Handle10.1073/pnas.96.19.10689 Occurrence Handle1:CAS:528:DyaK1MXmtFKjsr4%3D Occurrence Handle10485887
HJ Bussemaker D Thirumalai JK Bhattacharjee (1997) ArticleTitleThermodynamic stability of folded proteins against mutations Phys Rev Lett 79 3530–3533 Occurrence Handle10.1103/PhysRevLett.79.3530 Occurrence Handle1:CAS:528:DyaK2sXntFGntLo%3D
A Calcagnile T Basic-Zaninovic F Plombo E Dogliotti (1996) ArticleTitleMisincorporation rate and type on the leading and lagging strands of UV-damaged DNA Nucleic Acids Res 15 3005–3009 Occurrence Handle10.1093/nar/24.15.3005
C Claesson T Samuelsson F Lustig T Boren (1990) ArticleTitleCodon reading properties of an unmodified transfer RNA FEBS Lett 273 173–176 Occurrence Handle10.1016/0014-5793(90)81077-2 Occurrence Handle1:CAS:528:DyaK3MXjslyktw%3D%3D Occurrence Handle2226850
EC Cox C Yanofsky (1967) ArticleTitleAltered base ratios in the DNA of an Escherichia coli mutator strain Proc Natl Acad Sci USA 58 1895–1902 Occurrence Handle1:CAS:528:DyaF1cXislKltQ%3D%3D Occurrence Handle4866980
W Gilbert (1986) ArticleTitleThe RNA world Nature . 319–618
PA Gottlieb Y Prasad JB Smith AP Williams G Dinter-Gottlieb (1994) ArticleTitleEvidence that alternate foldings of the hepatitis delta RNA confer varying rates of self-cleavage Biochemistry 33 2802–2808 Occurrence Handle10.1021/bi00176a008 Occurrence Handle1:CAS:528:DyaK2cXhsFOntrY%3D Occurrence Handle8130192
S Griffiths-Jones A Bateman M Marshall A Khanna SR Eddy (2003) ArticleTitleRfam: An RNA family database Nucleic Acids Res 31 439–441 Occurrence Handle10.1093/nar/gkg006 Occurrence Handle1:CAS:528:DC%2BD3sXhvFSls7w%3D Occurrence Handle12520045
F Guo TR Cech (2002) ArticleTitleEvolution of Tetrahymena ribozyme mutants with increased structural stability Nat Struct Biol 9 855–861 Occurrence Handle1:CAS:528:DC%2BD38XosFCjsLY%3D Occurrence Handle12368901
KM Harrington IA Nazarenko DB Dix RC Thompson OC Uhlenbeck (1993) ArticleTitleIn vitro analysis of translational rate and accuracy with an unmodified tRNA Biochemistry 32 7617–7622 Occurrence Handle10.1021/bi00081a003 Occurrence Handle1:CAS:528:DyaK3sXks1Grs74%3D Occurrence Handle7688564
IL Hofacker W Fontana PF Stadler LS Bonhoeffer M Tacker P Schuster (1994) ArticleTitleFast folding and comparison of RNA secondary structures Monatsh Chem 125 167–199 Occurrence Handle10.1007/BF00818163 Occurrence Handle1:CAS:528:DyaK2cXivFaht7k%3D
KD James AD Ellington (1999) ArticleTitleThe fidelity of template-directed oligonucleotide ligation and the inevitability of polymerase function Orig Life Evol Biosph 29 375–390 Occurrence Handle1:CAS:528:DyaK1MXlvF2hsLo%3D Occurrence Handle10472627
EC Lai (2003) ArticleTitleRNA sensors and riboswitches: Self-regulating messages Curr Biol 13 R285–R291 Occurrence Handle1:CAS:528:DC%2BD3sXislChsLY%3D Occurrence Handle12676109
J Lambert N Moran (1998) ArticleTitleDeleterious mutations destabilize ribosomal RNA in endosymbiotic bacteria Proc Natl Acad Sci USA 95 4458–4462 Occurrence Handle1:CAS:528:DyaK1cXis1Ogsr0%3D Occurrence Handle9539759
M Levy AD Ellington (2001) ArticleTitleThe descent of polymerization Nat Struct Biol 8 580–582 Occurrence Handle1:CAS:528:DC%2BD3MXkvFCrs74%3D Occurrence Handle11427883
TM Lowe SR Eddy (1997) ArticleTitletRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence Nucleic Acids Res 25 955–964 Occurrence Handle1:CAS:528:DyaK2sXhvVahtrk%3D Occurrence Handle9023104
M Lynch (1996) ArticleTitleMutation accumulation in transfer RNAs: molecular evidence for Muller’s ratchet in mitochondrial genomes Mol Biol Evol 13 209–202 Occurrence Handle1:CAS:528:DyaK28XhtVylu7w%3D Occurrence Handle8583893
M Lynch (1997) ArticleTitleMutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes Mol Biol Evol 14 914–925 Occurrence Handle1:CAS:528:DyaK2sXlvVejurg%3D Occurrence Handle9287424
DH Mathews J Sabina M Zucker M Turner (1999) ArticleTitleExpanded sequence dependence of thermodynamic parameters provides robust prediction of RNA secondary structure J Mol Biol 288 911–940 Occurrence Handle1:CAS:528:DyaK1MXjtVers7Y%3D Occurrence Handle10329189
JS McCaskill (1990) ArticleTitleThe equilibrium partition function and base pair binding probabilities for RNA secondary structure Biopolymers 29 1105–1119 Occurrence Handle1:CAS:528:DyaK3cXkt1Snurk%3D Occurrence Handle1695107
EM Moody PC Bevilacqua (2003) ArticleTitleThermodynamic coupling of the loop and stem in unusually stable DNA hairpins closed by CG base pairs J Am Chem Soc 125 2032–2033 Occurrence Handle1:CAS:528:DC%2BD3sXnt1Krtg%3D%3D Occurrence Handle12590515
M Nakano EM Moody J Liang PC Bevilacqua (2002) ArticleTitleSelection for thermodynamically stable DNA tetraloops using temperature gradient gel electrophoresis reveals four motifs: d(cGNNAg), d(cGNABg), d(cCNNGg), and d(gCNNGc) Biochemistry 41 14281–14292 Occurrence Handle1:CAS:528:DC%2BD38XotlOmsLg%3D Occurrence Handle12450393
TV Pestova CU Hellen (2001) ArticleTitlePreparation and activity of synthetic unmodified mammalian tRNAi(Met) in initiation of translation in vitro RNA 7 1496–1505 Occurrence Handle1:CAS:528:DC%2BD3MXnvFSrsrk%3D Occurrence Handle11680854
DJ Proctor JE Schaak JM Bevilacqua CJ Falzone PC Bevilacqua (2002) ArticleTitleIsolation and characterization of a family of stable RNA tetraloops with the motif YNMG that participate in tertiary interactions Biochemistry 41 12062–12075 Occurrence Handle1:CAS:528:DC%2BD38XmvFKktL0%3D Occurrence Handle12356306
D Repsilber S Wiese M Rachen AW Schroder D Riesner G Steger (1999) ArticleTitleFormation of metastable RNA structures by sequential folding during transcription: time-resolved structural analysis of potato spindle tuber viroid (−)-stranded RNA by temperature-gradient gel electrophoresis RNA 5 574–584 Occurrence Handle1:CAS:528:DyaK1MXisVCisLo%3D Occurrence Handle10199573
E Rocha A Danchin (2002) ArticleTitleBase composition bias might result from competition for metabolic resources Trends Genet 18 291–294 Occurrence Handle1:CAS:528:DC%2BD38XktVCku7Y%3D Occurrence Handle12044357
K Salehi-Ashtiani JW Szostak (2001) ArticleTitleIn vitro evolution suggests multiple origins for the hammerhead ribozyme Nature 414 82–84 Occurrence Handle1:CAS:528:DC%2BD3MXot1aisbk%3D Occurrence Handle11689947
JR Sampson OC Uhlenbeck (1988) ArticleTitleBiochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro Proc Natl Acad Sci USA 85 1033–1037 Occurrence Handle1:CAS:528:DyaL1cXkvVOhs7c%3D Occurrence Handle3277187
T Schmitt N Lehman (1999) ArticleTitleNon-unity molecular heritability demonstrated by continuous evolution in vitro Chem Biol 6 857–869 Occurrence Handle1:CAS:528:DyaK1MXotVygsrw%3D Occurrence Handle10631514
EA Schultes DP Bartel (2000) ArticleTitleOne sequence, two ribozymes: Implications for the emergence of new ribozyme folds Science 289 448–452 Occurrence Handle1:CAS:528:DC%2BD3cXlsV2gtLg%3D Occurrence Handle10903205
Z Shu PC Bevilacqua (1999) ArticleTitleIsolation and characterization of thermodynamically stable and unstable RNA hairpins from a triloop combinatorial library Biochemistry 38 15369–15379 Occurrence Handle1:CAS:528:DyaK1MXmslCksro%3D Occurrence Handle10563823
JB Smith PA Gottlieb G Dinter-Gottlieb (1992) ArticleTitleA sequence element necessary for self-cleavage of the antigenomic hepatitis delta RNA in 20 M formamide Biochemistry 31 9629–9635 Occurrence Handle1:CAS:528:DyaK38XlvVGrt7Y%3D Occurrence Handle1390740
M Szymanski MZ Barciszewska J Barciszewski VA Erdmann (2000) ArticleTitle5S ribosomal RNA database Y2K Nucleic Acids Res 28 166–167 Occurrence Handle1:CAS:528:DC%2BD3cXhvVKjsLc%3D Occurrence Handle10592212
K Takai H Takaku S Yokoyama (1996) ArticleTitleCodon-reading specificity of an unmodified form of Escherichia coli tRNA1Ser in cell-free protein synthesis Nucleic Acids Res 24 2894–2899 Occurrence Handle1:CAS:528:DyaK28XlsFChur4%3D Occurrence Handle8760870
E van Nimwegen JP Crutchfield MA Huynen (1999) ArticleTitleNeutral evoltuion of mutational robustness Proc Natl Acad Sci USA 96 9716–9720 Occurrence Handle1:CAS:528:DyaK1MXlsVymtrk%3D Occurrence Handle10449760
M Vendruscolo A Maritan JR Banavar (1997) ArticleTitleStability threshold as a selection principle for protein design Phys Rev Lett 78 3967–3970 Occurrence Handle1:CAS:528:DyaK2sXjtleku7s%3D
GP Wagner G Booth H Bagheri-Chaichian (1997) ArticleTitleA population genetic theory of canalization Evolution 51 329–347
A Walter D Turner J Kim M Lyttle P Müller D Mathews M Zuker (1994) ArticleTitleCoaxial stacking of helices enhances binding of oligoribonucleotides Proc Natl Acad Sci USA 91 9218–9222 Occurrence Handle1:CAS:528:DyaK2MXhtFWksbc%3D Occurrence Handle7524072
S Wuchty W Fontana IL Hofacker P Schuster (1999) ArticleTitleComplete suboptimal folding of RNA and the stability of secondary structures Biopolymers 49 145–165 Occurrence Handle1:CAS:528:DyaK1MXpsFOqsQ%3D%3D Occurrence Handle10070264
Acknowledgments
The authors thank Walter Fontana and Rob Knight for technical advice and Kourosh Salehi-Ashtiani and Jack Szostak at Mass General Hospital for providing selected ribozyme sequences. This work was supported in part by the Santa Fe Institute and grants from the NSF (Grant DEB-0303636) to L.A.M., grants from the NSF (Grant EIA-0218447) and the NIH–NIBIB (Grant 8R01EB002043) to A.D.E., and NSF-IGERT fellowships in computational phylogenetics to J.F.L and M.C.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meyers, L.A., Lee, J.F., Cowperthwaite, M. et al. The Robustness of Naturally and Artificially Selected Nucleic Acid Secondary Structures. J Mol Evol 58, 681–691 (2004). https://doi.org/10.1007/s00239-004-2590-2
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2590-2