
Abstract
The mutational mechanism underlying the striking diversity in MHC (major histocompatibility complex) genes in vertebrates is still controversial. In order to evaluate the role of inter- and intragenic recombination in MHC gene diversification, we examined patterns of nucleotide polymorphism across an exon/intron boundary in a sample of 31 MHC class IIB sequences of three-spined stickleback (Gasterosteus aculeatus). MHC class IIB genes of G. aculeatus were previously shown to be under diversifying (positive) selection in mate choice and pathogen selection experiments. Based on recoding of alignment gaps, complete intron 2 sequences were grouped into three clusters using maximum-parsimony analysis. Two of these groups had >90% bootstrap support and were tentatively assigned single locus status. Intron nucleotide diversity within and among loci was low (p-distance within and among groups = 0.016 and 0.019, respectively) and fourfold lower than the rate of silent mutations in exon 2, suggesting that noncoding regions are homogenized by frequent interlocus recombination. A substitution analysis using GENECONV revealed as many intergenic conversion events as intragenic ones. Recombination between loci may explain the occurrence of sequence variants that are particularly divergent, as is the case in three-spined stickleback, with nucleotide diversity attaining dN = 0.39 (peptide-binding residues only). For both MHC class II loci we also estimated the amount of intragenic recombination as population rate (4Ner) under the coalescent and found it to be approximately three times higher compared to point mutations (Watterson estimate per gene, 4N e μ). Nonindependence of molecular evolution across loci and frequent recombination suggest that MHC class II genes of bony fish may follow different evolutionary dynamics than those of mammals. Our finding of widespread recombination suggests that phylogenies of MHC genes should not be based on coding segments but rather on noncoding introns.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MHC (major histocompatibility complex) genes are prime examples of genes under diversifying (=positive) selection. Being the most polymorphic loci identified in vertebrate species thus far, MHC class I and II gene loci often display as many as several hundred alleles (Parham and Ohta 1996). Due to their crucial role in presenting pathogen-derived peptides to specialized cells of the immune system, a role of diversifying selection was proposed in 1975 (Doherty and Zinkernagel 1975). According to the “heterozygote advantage hypothesis,” MHC-heterozygous individuals recognize a wider array of foreign peptides, ultimately resulting in higher Darwinian fitness in the face of pathogen infections. An alternative, nonexclusive form of diversifying selection is frequency-dependent selection, in which individuals carrying rare alleles have a selective advantage under host–pathogen coevolution (Takahata and Nei 1990). Analyses of DNA sequence polymorphism found substantially elevated rates of nucleotide changes resulting in the replacement of an amino acid relative to silent mutations, in line with the above hypotheses of positive (=diversifying) selection (Hughes and Nei 1988).
Since then, experimental work supported molecular inferences. In mice and fish both sexual selection (Potts et al. 1991; Reusch et al. 2001a) and natural selection by parasites and pathogens (Langefors et al. 2001a; Penn et al. 2002; Wegner et al. 2003a,b) maintain high allelic polymorphism within populations. While this work explains the maintenance of MHC polymorphism given a diverse array of alleles, the ultimate mutational mechanism that generates more than 100 sequence variants at classical MHC loci is less clear (Martinsohn et al. 1999). It was originally posited that intralocus variation generally originates from point mutations (Klein 1986). However, if point mutations are the only generator of genetic variation in MHC genes, then the mutation rate should be extremely high, which has not been supported by sequence analyses (Satta et al. 1993). Alternatively, the divergence time between MHC alleles must be extremely long, with allelic lineages predating speciation (Klein 1986). Such “trans-species evolution” of MHC genes is most frequently found among mammalian species (Kupfermann et al. 1992; Kriener et al. 2000) but was subsequently also reported in fish (Figueroa et al. 2000) and birds (Sato et al. 2001).
While under the assumption of very old allele ages, point mutations would suffice for explaining allele diversity and divergence. The mosaic pattern among MHC genes consisting of a complex assortment of various sequence motifs suggested that the diversification of MHC sequence variants may also be a result of recombination sensu lato (Parham and Ohta 1996; Martinsohn et al. 1999). Possible mechanisms include gene conversion-like events and (repeated) microrecombination. Since many classical MHC genes occur as clusters of functionally intact, duplicated genes, interlocus recombination through unequal crossing-over may also generate sequence polymorphism (Ohta 1999). Throughout this paper, we do not distinguish between gene conversion and recombination. While the molecular mechanism of sequence exchange may be different, the observed pattern of polymorphism is similar for sequence data of limited length typically available in population data sets (Wiehe et al. 2000; Richman et al. 2003).
According to the “birth-and-death model” of MHC evolution (Nei and Hughes 1992; Gu and Nei 1999), MHC loci are duplicated repeatedly and maintained in the populations for long times, whereas others have lost their functionality. Models have shown that locus duplication under intergenic recombination may interact synergistically and lead to a higher sequence diversity in terms of particularly divergent sequence variants (Ohta 1999). Divergent alleles, in turn, may confer fitness advantages under the divergent allele advantage (Wakeland et al. 1990; Richman et al. 2001, 2003), which is when heterozygous individuals carrying very different copies have a higher fitness than heterozygotes with little divergence.
Considerable progress has been made in detecting gene conversion from samples of DNA sequences (Sawyer 1999; McVean et al. 2002; Posada 2002; Stumpf and McVean 2003), and one hope is that the improvement in bioinformatic methods may improve understanding of MHC evolution. A major new development is the application of coalescent theory (Hudson and Kaplan 1985) for predicting recombination rates from a sample of DNA sequences (Brown et al. 2001; Hudson 2001; Stumpf and McVean 2003). This way, not only the presence of recombination, but also its relative contribution can be estimated, substantially increasing qualitative notions of recombination/gene conversion in earlier analyses of sequence polymorphism (Parham and Ohta 1996; see further examples in Martinsohn 1999).
The goal of this paper is to explore and quantify the importance of recombination/gene conversion in relation to point mutations in shaping the diversification of MHC class IIB genes in populations of three-spined sticklebacks (Gasterosteus aculeatus). The MHC class IIB genes of this species currently build one of the most convincing cases for pathogen defense genes that are under diversifying selection. In both sexual and pathogen selection experiments, a crucial role for stickleback MHC class IIB genes in conferring resistance to pathogens and attractiveness to potential mating partners has been demonstrated (Reusch et al. 2001a; Wegner et al. 2003a, b). In addition, three-spined stickleback are increasingly becoming a model of evolutionary biology, including genomics (Reusch et al. 2004), calling for a closer analysis of those genes that have proven to play an important role in parasite defense and mate choice. An analysis of sequence evolution in nonmammalian taxa with respect to potential recombination is rare (but see Langefors et al. 2001b) but warranted given that the evolution of MHC genes may differ considerably among vertebrate classes (Edwards et al. 1995; Hess and Edwards 2002; Stet et al. 2003).
Materials and Methods
Sampling, Sequence Acquisition, and Molecular Methods
Sequence data were obtained by cloning and sequencing MHC class IIB sequences in DNA samples of 48 individuals of Gasterosteus aculeatus from five freshwater sites in Schleswig-Holstein, Germany, and one stream site in British Columbia, Canada. The Canadian population is estimated to have diverged from the European ones between 200,000 and 500,000 years ago based on mtDNA polymorphism (Orti et al. 1994). The sites in northern Germany were no more than 50 km distant from each other. We sequenced the polymorphic 210-bp region of exon 2, encompassing residues 25–94 of the human MHC class II β-chain (Brown et al. 1993) plus the entire intron 2. Exon 2 encodes parts of the functionally important peptide-binding region (PBR) of MHC class II molecules. For obtaining PCR amplicons prior to cloning, we used primers situated at the 5′ end of exon 2 and in the first 22 bp (base pairs) of the conserved exon 3 (GA_MHCII_exon3start: 5′-CGT CTC AGA GTG CAG CCT GAC GT-3′ [K.M. Wegner, unpublished]). The upstream primer (GA11, 5′-AAC TCC ACT GAG CTG AAG GAC ATC-3′ [Sato et al. 1998]) anneals at the 5′ end of exon 2, a region that is conserved in several bony fish. PCR conditions were as follows: a 100-μL reaction contained 5 units of high-fidelity Taq (Roche), 1× reaction buffer, 1.5 mM MgCl2, a 200 μM concentration of each dNTP, 0.1% (wt:vol) bovine serum albumin, and approximately 1–10 ng of genomic DNA as template. The PCR program was: 3-min initial denaturing at 94°C, 30–32 cycles of 15-s denaturing at 94°C, 1-min annealing at 58°C, 4-min extension at 72°C, and 7-min final extension at 72°C. The net amplicon size varied between 1031 and 1730 bp, with on average 3.4 (range, 2–5) distinct bands per fish. Note that the different lengths of PCR amplicons do not correspond to loci, or to unique alleles (Reusch et al. 2004; this paper). Not all amplicon sizes were present in every fish, suggesting that there is substantial interindividual variation in the number and presence of individual MHC class IIB loci. PCR amplification products were excised from a 1.5% agarose gel, purified, ligated, and cloned into competent E. coli using the TopoTA-cloning kit (Invitrogen). Between 6 and 12 transformant colonies per band were picked and their plasmid DNA was prepared using a plasmid preparation kit (Qiagen, Hilden, Germany). Inserts were sequenced forward and reverse on an ABI 3100 automated sequencer, using a long-read capillary (80 cm), standard M13 forward and reverse primers, and the BigDye 3.1 sequencing kit (Applied Biosystems, Weiterstadt, Germany). In total, our data set comprised 3601 readable sequence runs >850 bp. We obtained complete intron 2 sequences in 31 MHC class IIB sequences. These sequences have been submitted to GenBank (accession numbers DQ016399–DQ016429; alignment, supplementary Table S1).
On average, we found 4.5 sequence variants per individual fish (maximum, 8), excluding a putative pseudogene with very divergent exon and intron sequences that was present in almost every fish analyzed (T.B.H. Reusch, unpublished data). Individual fish may thus possess between two and four functional MHC class IIB gene loci. We repeated cloning and sequencing in eight individuals to examine the reproducibility of cloning and sequencing. PCR artifacts may potentially create a pattern that will exactly resemble gene recombination in nature (Bradley and Hillis 1997). We found that most sequence variants were reproducible. In cases of nonidentical sets of sequences in both cloning runs, the additional MHC class IIB sequence clearly did not originate from a PCR artifact based on the distribution of shared and nonshared sequence motifs. Nevertheless, as a precaution against PCR artifacts during cloning, the majority of sequence variants (25/31) were obtained at least twice independently in different individuals.
We based our sequence characterization on genomic DNA because we wished to analyze nucleotide polymorphism and recombination patterns across the exon 2/intron 2 boundary, increasing the power for detecting recombination (Hughes 2000). We tested for the expression of the sequences by SSCP (single-strand conformation polymorphism) as described in Binz et al. (2001) and Reusch et al. (2001a). Genotyping was done in a subsample of individuals from which we simultaneously obtained cDNA and genomic DNA as a PCR template. We found that 14 of 16 identified signals were present in both cDNA and genomic DNA as template (Wegner et al. 2003a, b).
Identification of Putative MHC Class IIB Gene Loci
Sticklebacks are estimated to possess up to six different MHC class IIB gene loci (Sato et al. 1996; Reusch et al. 2001a). A previous analysis using a large insert library revealed that at least two of these loci reveal less than 2% sequence divergence in the introns and have originated by very recent gene duplication (<2 MYA) (Reusch et al. 2004). Nevertheless, we attempted to identify sequence groups indicative of loci by examining the sequence polymorphism of the entire intron 2 by taking advantage of the information contained in the alignment gaps (i.e., indels). Indels of noncoding sequences may evolve faster than point mutations (Britten et al. 2003). In order to utilize information contained in indels, we used maximum parsimony analysis (MP) because this method allows for recoding alignment gaps as parsimony informative sites (Lee 2001). Prior to the analysis, sequences were aligned using the ClustalW algorithm, in combination with manual alignment. MP analysis was conducted using MEGA3 (Kumar et al. 2004), with 450 initial trees and under the close neighbor interchange (CNI) algorithm with a search factor of 3. The robustness of the resulting tree was assessed in 250 bootstrap runs. For gap coding we used the fragment coding approach (Lee 2001). All alignment gaps were recoded as present or absent (if there were no additional polymorphisms in the remaining sequence portions), or as multistage characters if there were additional substitution polymorphism in the stretches with nucleotides. In this way, we obtained 38 recoded state characters in addition to 32 parsimony informative sites consisting of point substitutions to be included in the MP analysis.
We also used the information in exon 2 to construct a dendrogram based on inferred amino acid composition (neighbor-joining algorithm based on p-distance, Poisson corrected, implemented in MEGA3). This was done for comparative purposes, as most MHC sequence data sets use exon 2 to resolve allelic lineages, while sequence information from noncoding introns is often unavailable. We were particularly interested whether the tree topology based on coding exon 2 or noncoding intron 2 would be similar or different, as the latter outcome would indicate recombination across the exon–intron boundary. Also, it is qualitatively predicted that under frequent recombination, phylogenetic trees lack deep allelic lineages (Gu and Nei 1999).
Analyses of Sequence Polymorphism and Positive Selection
First, we quantified the sequence diversity among synonymous and nonsynonymous sites within the coding region (exon 2) using the software MEGA3 (Kumar et al. 2004). We tested for positive (diversifying) selection by computing the number of nonsynonymous substitutions per nonsynonymous site (dN) with the silent substitution rate (dS). This analysis was performed separately for all 70 codons of exon 2 and for the 21 putative peptide-binding codons according to the crystalline structure of the human MHC molecule (Brown et al. 1993). The excess of dN over dS was statistically examined using a Z-test (Hughes and Nei 1988).
Second, we compared the silent polymorphism in exon 2 to the total nucleotide polymorphism in intron 2. In MHC genes, recombination across an intron–exon boundary will lead to very different patterns of polymorphism. Strong diversifying selection in exon 2 will simultaneously increase the polymorphism at silent sites. In contrast, in the adjacent intron, recombination will tend to make sequences more similar under selective neutrality. Therefore the nucleotide polymorphism will be decreased in introns relative to the synonymous substitution rate in exon 2 (Hughes 1999, 2000). Note that this does not imply that the recombination patterns vary between exonic and intronic portions of the gene (Hughes 1999). If intergenic exchange is also common, this will lead to homogenization of intron sequences across putative gene loci (Hughes 1999; Ohta 1999).
Detection of Recombination
We used three computer programs to detect recombination events. The program GENECONV (Sawyer 1999) employs a substitution model that scans for significant clustering of substitutions. Clusters are tested against a null hypothesis by permutation (10,000 runs). This method has a high statistical power of detecting gene conversion when it is actually present, while the risk of obtaining false positive results is low (Brown et al. 2001; Posada 2002). A global p-value that is adjusted for the number of comparisons is calculated, as well as a Bonferroni-adjusted p-value for each recombination event that was detected between any pair of sequences.
In order to evaluate the relative amount of recombination (intragenic only) in comparison to point mutation rate, we analyzed the genealogy of a sample of MHC class IIB sequences using a modification of the coalescent method of Hudson (2001), implemented in the software LDHAT (McVean et al. 2002). LDHAT estimates recombination using an importance sampling approach (Fearnhead and Donnelly 2001) of the genealogy of a sample of DNA sequences. Subsequently, a likelihood function is fitted, taking into account recurrent mutations and a finite population size. The inclusion of recurrent mutation is critically important when examining MHC genes that are under strong diversifying selection (Richman et al. 2003). The amount of population recombination is quantified as ρ = 4N e r (Hudson and Kaplan 1985) and tested by permutation for statistical difference from zero. The 4N e r value can be compared to the (point) mutation rate that is estimated in a finite sites model according to the Watterson (1975) estimate 4N e μ in order to quantify the relative contribution of recombination and point mutation in generating sequence diversity (McVean et al. 2002; Richman et al. 2003). The robustness of the coalescent model to symmetric balancing selection, and hence its applicability to MHC data, was recently verified by Richman and coworkers (2003) using simulations. Also, 4N e r/4N e μ has been shown to be robust against biases in the estimation of the per-site mutation rate (cf. Table 1, McVean et al. 2002). Note that values of ρ >100 cannot be estimated with confidence in LDHAT.
We used the recombination analyses provided in DNASP ver. 3.99 (Rozas et al. 2003), which calculates the minimum number of recombination events, ρM, according to Hudson and Kaplan (1985). DNASP was also used to locate the sites where putative recombination events took place.
Results
Maximum Parsimony Analysis of MHC Class IIB Intron 2 Sequences
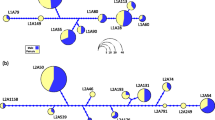
All 31 sequences could be unambiguously aligned (supplementary information, Table S1). In accordance with two complete MHC class IIB genes previously identified in a large insert library (Reusch et al. 2004), 22 of 31 sequence variants contained a 10-mer tandem repeat with the consensus sequence CCTTGTAGAA (see supplementary information, Table S1; base position 1138 ff). This repeat region is responsible for most of the length polymorphism observed in intron 2. This repetitive region was excluded from further analysis of nucleotide polymorphism because such microsatellites will have higher mutation rates and evolve under a different mutation model (stepwise mutation model) than nonrepetitive sequences. In order to improve the alignment we introduced a total of 25 alignment gaps, none of which was ambiguous. Maximum-parsimony analysis based on a combination of point substitutions and recoded gap characters revealed three major sequence groups, denoted A through C (Fig. 1), two of which (B and C) had >90% bootstrap support (Fig. 1). The sequence variants in groups B and C were therefore denoted Gaac-DCB*01-09 and Gaac DDB*01-06 according to the suggested MHC nomenclature. Group A contained two sequence variants (Gaac-DAB and -DBB) that were previously identified to be independent MHC class IIB loci using a large insert library (Reusch et al. 2004). Hence, this group must be composed of at least two independent MHC class IIB loci. The sequence variants from the unresolved group A are denoted Gaac-DXB, except for the previously identified Gaac-DAB and -DBB. Note that the phylogenetic separation of intron 2 depends entirely on the recoded state characters of indels. If omitted, neither MP nor neighbor joining is able to produce statistically supported sequence groups.

Major histocompatibility class IIB genes in three-spined sticklebacks (Gasterosteus aculeatus): bootstrap consensus tree (majority rule) of a maximum parsimony analysis of single nucleotide polymorphisms and indels of 31 complete intron 2 sequences. Note that group A must consist of more than one gene locus because the variants Gaac-DAB and –DBB were previously identified as independent loci in a large insert library. The analysis was carried out in MEGA3 (Kumar et al. 2004) using the CNI algorithm (search factor =3) and 450 initial trees. Only bootstrap values >40% are given.
For comparison with the MP analysis based on the second intron, we also constructed a phylogenetic tree based on the inferred amino acid composition of the second exon (p-distance, Poisson corrected) using the neighbor-joining algorithm implemented in MEGA3. The topology of the resulting bootstrap consensus was very different compared to the second intron. The sequence groups A through C identified using intron 2 polymorphism are distributed among several separate branches of the NJ tree, some of which have >80% bootstrap support at the branch tips, i.e., comprising two or three sequence variants only. The same qualitative result applied to a MP analysis of amino acid or base composition of exon 2 (data not shown).
Sequence Polymorphism in Coding and Noncoding Portions of MHC Class IIB Genes
Our sample of MHC class IIB genes may have been biased if some sequence types are too divergent to be amplified by our cloning primers. We therefore checked for a relationship between the chances of sequences to be amplified, which should be proportional to the mean sequence divergence in a genotype, and the number of sequences we recovered from each fish. Specifically, we computed the mean sequence divergence for all exon 2 positions using the Kimura two-parameter and a transition:transversion ratio of 2 for each fish genotype that was sequenced to near-saturation (i.e., >10 clones per band, on average 34 sequences per fish). In this sample of 30 fish, we find no correlation between the mean sequence divergence and the number of unique sequence variants that could be recovered (r2 = 0.03, p = 0.03).
We then examined sequence polymorphism for each of the three MHC class IIB sequence groups separately. Sequence diversity within the second exon was high, with 69 of 210 (33%) polymorphic sites. The average distance (p-distance ± SE, Poisson corrected) in terms of inferred amino acid sequences within groups A, B, and C ranged from 0.18±0.03 to 0.24±0.04. The magnitude of replacement mutations in putative codons of the PBR (peptide-binding region) was particularly high and attained a value of dN = 0.42 in sequence group A (Table 1). Differences between dN and dS throughout all three sequence groups were highly significant in a Z-test implemented in MEGA3, indicating positive selection. The majority of the polymorphic residues (19/21=90%) were found at positions identical to those involved in antigen binding of human MHC class IIB genes (data not shown; Brown et al. 1993).
The polymorphism of the intron 2 sequences was very different. Here, we found low nucleotide diversity, on average four times lower than the synonymous substitution rate within exon 2. This difference is statistically different in all three identified sequence groups given the nonoverlapping confidence intervals (Table 1). Interestingly, the between-group polymorphism in the second intron (p-distance±1SE) was very low (group A vs. B, 0.0154±0.002; group A vs. C, 0.019±0.003; group B vs. C, 0.0185±0.003). Accordingly, the amount of polymorphism hardly increases when pooling all three groups (common p-distance = 0.016). This highlights the similarity of the intron 2 sequences outside the alignment gaps. Moreover, this finding stresses the essential information contained in the indels for uncovering the relatedness among intron 2 types using maximum parsimony.
Presence and Relative Amount of Recombination
All three methods detected statistically significant recombination. The substitution approach implemented in GENECONV revealed low statistical p-values for the global tests (N = 31 sequences; 10,000 permutations, p < 0.001). In addition to a significant global test, GENECONV detected 22 pairwise recombination events that were significant at a Bonferroni-adjusted significance level of α ≤ 0.05. Note that this estimate is probably conservative because the large number of pairwise comparisons requires a correction of the nominal statistical α-value by dividing through 465. Of 31 sequences, 15 (=48%) were involved in at least one pairwise recombination event. We then examined which pairs of MHC sequences were involved in statistically significant recombination based on the groupings obtained by MP analysis. Half (11/22) of these events occurred within, and the other half between, putative MHC class IIB loci. Interestingly, no intralocus recombination was detectable within group B sequences, while eight exchanges of B with group A sequences were detected, all of which must be intergenic. Of four recombination events involving Canadian sequence variants, three involved one European sequence type.
The coalescent approach for detecting recombination is valid only when applied to single locus data. Therefore, we restricted this analysis to the sequence variants belonging to intron 2 groups B and C, the strongly supported groups of the MP analysis. Under the coalescent approach implemented in LDHAT, we found that the amount of population recombination ρ= 4N e r exceeded the role of point mutations θ=4N e μ 2.8-fold in one putative gene locus (B) and 3.3-fold in the other (Table 2). We also examined the robustness of the high amount of 4N e r identified in our data using 0.5× or 2× the estimate of Watterson’s θ. For both sequence groups, results remained at the maximal estimable value of 4N e r = 100.
The approach in DNASP recognized a minimum (ρM) of 5–30 recombination events according to Hudson and Kaplan (1985) in the three sequence groups, the majority of which involved exon 2 (Table 3).
Discussion
Recombination may reduce or promote genetic polymorphism depending on the selection regime of the target gene segment (Nei and Lee 1980; Strohbeck 1983; Hughes 1999). Under directional selection or neutrality, recombination will lead to gene homogenization and concerted evolution. However, just the opposite effect may be observed under diversifying (positive) selection (Hughes 2000). MHC genes are prime examples to study contrasting effects of recombination because two opposing selective regimes shape the genetic polymorphism: positive selection in the second exon and neutrality in the second intron. The data set here was obtained from sticklebacks, one of the few species where diversifying selection has also been experimentally verified in mate choice and parasite selection experiments (Reusch et al. 2001; Wegner 2003a, b), strengthening any conclusions solely based on molecular genetic data.
Among a sample of 31 sequence variants, we found a fourfold higher substitution rate at synonymous sites in exon 2 compared to the adjacent intron 2. This is the pattern expected under gene conversion, where homogenization of introns relative to the synonymous substitutions in the exon will decrease the amount of sequence difference in this region (Hughes 1999, 2000; Ohta 1999). This finding alone is not sufficient for demonstrating gene conversion across loci but could simply be explained by a long-standing balanced polymorphism that is segregating within a single gene (e.g., Kreitman and Hudson 1991). However, the extremely limited polymorphism between putative MHC class II B loci (p-distance between groups ≤0.019) can be accounted for only if frequent intergenic exchange is assumed.
In support of this notion, we found statistically highly significant signals of intra- and intergenic recombination using two independent methodologies in populations of three-spined stickleback that may explain the observed differences in neutral polymorphism among coding and noncoding regions. Results obtained by a coalescent approach (LDHAT) suggest that the effects of intragenic recombination alone on MHC sequence polymorphism are approximately three times higher than the role of point mutations (4Neμ). In addition, we find that at least half of the total recombination detected in all sequence variants using a substitution approach is due to intergenic recombination events. This qualitatively doubles the role of recombination (intra- and intergenic) for generating MHC sequence polymorphism. The data set comprises sequence variants from Europe and Canada that diverged 2–5 × 105 years ago (Orti et al. 1994). While the sample size of the Canadian fish is not very high, we qualitatively conclude from three recombination events detected between European and Canadian MHC sequences that some of the polymorphism in MHC class IIB is due to relatively old recombination events. This, however, does not rule out a dominant role for recombination for the rapid generation of novel allelic variation (Parham and Ohta 1996).
Since our data set comprised mostly MHC class IIB sequences that were at least twice independently confirmed in other individuals, PCR or cloning artifacts, for example, through Taq-polymerase template switching (Bradley and Hillis 1997), are probably not responsible for the high amount of recombination detected. The six sequence variants that were found only once are not involved more often in recombination than expected (4/22 pairwise recombination events detected in GENECONV). Nevertheless, our results may have been biased since we excluded many rare sequence variants that occurred only once. To examine this, we resampled 31 sequences from a total number of 64 distinct exon 2 sequences that are available from the 48 analyzed fish (singletons plus confirmed sequence variants in more than one individual). We repeated the analysis in GENECONV with 10 such resampling rounds. All data sets revealed highly significant signals of recombination (all global p’s < 0.001, 10,000 randomizations). In the pairwise analysis, we found on average a somewhat larger number of recombination events (26.9) in the resampled data sets, suggesting that we may have underestimated the true amount of recombination.
In order to detect recombination in a substitution approach, the homology criterion within a sample of sequence variants may be orthology or paralogy (Posada 2002). Thus, the results obtained using the substitution approach implemented in GENECONV are reliable, regardless of whether sequence variants were sampled from one or several loci. In contrast, the coalescent approach implemented in LDHAT and DNASP in order to obtain population estimates of the amount of recombination requires that all sequence variants come from one locus, which we achieved after MP analysis of the entire intron 2. Additional simplifying assumptions such as constant population size and no genetic structure also need to apply (Hudson 2001). Simulations by Fearnhead and Donnelly (2001) have revealed that the quotient of ρ/θ as an estimate of the relative amount of recombination (ρ) compared to point mutations (θ) is robust against several violations of the underlying coalescent model, a panmictic population of constant size. First, any demographic dynamic that increases or decreases the effective population size (Ne) cancels out in 4N e r/4N e μ. Geographic population structure up to a fixation index (FST) of 0.2 was shown to yield rather conservative estimates of ρ/θ. FST in the stickleback populations investigated in this study has formerly been found to be around FST=0.2 (Reusch et al. 2001b). Hence, the population substructure should not cause any problem in our analyses.
Two MHC class IIB sequence variants included in this data set were previously identified in a 100-kbp contiguous genomic segment. Hence, they belong unequivocally to two different loci that occur in tandem arrangement approximately 27 kbp distant from one another (Gaac-DAB and Gaac-DBB [Reusch et al. 2004]). Among the two specific alleles they carry, we could detect a pairwise recombination event (GENECONV, pairwise p = 0.007, Bonferroni correction applied) that by definition must be intergenic. In the larger sequence sample presented here, this isolated finding of interlocus recombination was confirmed.
Recombination between loci effectively decreases the phylogenetic signal that may be used for locus identification. Among the sequences analyzed here, the overall sequence similarity in the second intron was remarkably high (p-distance among all 31 sequences = 0.016), although the sample probably consists of four different class IIB loci. Had this been a sequence sample of any mammalian MHC gene, such a high similarity would indicate identical MHC class II locus affiliation. For example, in the human DRB locus, the intron divergence between allelic lineages of the same gene locus is three to four times larger than in sticklebacks (d = 0.06–0.08 [Bergström et al. 1998]). This result implies that at least some of the studied loci are the product of recent gene duplication. Alternatively, the locus duplication may be old, yet ongoing recombination homogenizes the introns such that similarities prevail. At least for the loci Gaac-DAB and -DBB the sequence polymorphism suggests that the duplication was indeed recent because the intron divergence is constant along the entire gene, while we would expect decreasing homogenization (hence more divergence) away from the site of strong positive selection, exon 2 (Reusch et al. 2004). To examine the relative role of ongoing intergenic recombination and the time since gene duplication, longer contiguous stretches of the MHC region are clearly needed (Wiehe et al. 2000).
If interlocus recombination is a major force behind MHC polymorphism, then alleles from the same locus would not form a monophyletic cluster. Rather, alleles from different loci intermingle into a bushlike dendrogram (Gu and Nei 1999), and this is what a phylogenetic analysis based on the second exon revealed (Fig. 2). Positive selection in combination with frequent recombination completely erases any phylogenetic signal from the portion of the gene most critical to immune function, exon 2. Therefore, we consider the tree presented in Fig. 2 solely as a heuristic tool to examine sequence similarities, without any further phylogenetic inferences.

Relationship among 31 MHC class IIB sequences in three-spined sticklebacks. A neighbor-joining tree was constructed in MEGA3 (Kumar et al. 2004; p-distance, Jukes–Cantor correction) of inferred amino acid sequences of the polymorphic exon 2. Affiliations with groups based on intron 2 maximum parsimony analysis are given as capital letters by the brackets. Only bootstrap values >40% are shown (250 runs).
Deep allelic lineages suggest that intergenic exchange in MHC class II genes of mammals is rare (Gu and Nei 1999). The only other quantitative analysis of gene conversion in a nonmammalian species was conducted in a sequence sample of Atlantic salmon, a species that possesses only one class IIB locus (Langefors et al. 2001b) and cannot undergo intergenic recombination. The importance of intragenic recombination in MHC class II genes has recently been stressed in deer mouse (Richman et al. 2003), where it contributes 12-fold more to sequence diversity than point mutations. For mammalian class II genes, experimental studies in sperm of mice and men revealed that the rate of gene conversion is surprisingly high and exceeds the rate of point mutations by three to four orders of magnitude (Högstrand and Böhme 1994; Zangenberg et al. 1995). In light of this experimental evidence, it is surprising that many authors still ignore the potential role of recombination. In order to explain some of the common motifs shared among MHC alleles, convergent evolution has frequently been invoked as an alternative explanation (Kriener et al. 2000; Figueroa et al. 2000). Under convergence, the same blocks substitutions are found at identical sites in two or more independent alleles due to similar selection pressures. Yet, to produce shared motifs found in different MHC alleles, an enormous number of random substitutions is required. This, is turn, requires an extraordinarily high mutation rate, or a very long time period. Several authors have shown that the mutation rate is not higher at MHC loci than at other genes (e.g., Satta et al. 1993). Yeager and Hughes (1999) estimated that for two unrelated ancestral sequences sharing an amino acid sequence motif of three residues, time intervals >20 MYA are required. Clearly invoking convergence rather than evolution by recombination/gene conversion view requires many more unrealistic assumptions.
MHC sequence evolution including longer stretches of noncoding gene segments has been studied in few other natural populations, and we are not aware of any study in fish. This is clearly a shortcoming, given that an analysis of paralogous and orthologous relationships among gene families such as MHC should rather be undertaken using noncoding segments of the gene (Bergström et al. 1998; Hughes 2000; Elsner et al. 2002). Phylogenetically supported clusters of exon 2 sequences, encompassing the peptide-binding region, have often been interpreted as evidence against a prominent role of recombination in shaping MHC variation (e.g., Kupfermann et al. 1992; Figueroa et al. 2000; Kriener et al. 2000; Sato et al. 2001). However, under intra- and intergenic recombination, the detection of phylogenetic branches under a point-mutation model is flawed and, therefore, cannot be used for identifying the homologous relationships among gene loci.
The evolution of MHC genes in sticklebacks, and possibly other fish species (Stet et al. 2003), may be remarkably different from that in well-studied mammalian species. In fact, two main features make MHC evolution in three-spined sticklebacks more similar to that found in birds than to that in mammals. First, the absence of locus-specific clustering of exon 2 and intron 2 and, second, the large variations in haplotypic gene number (Edwards et al. 1995; Hess and Edwards 2002; Westerdahl et al. 2000) markedly contrast with well-known mammalian examples. Two nonexclusive evolutionary models of MHC evolution may explain the lack of interlineage allelic divergence, the “recent duplication model” and the “gene conversion model” (Hess and Edwards 2002). The gene conversion model of evolution of the stickleback MHC loci is supported by differences in nucleotide diversity between transcribed and nontranscribed portions of the MHC class IIB genes, and by the frequency of recombination between more distantly related sequence variants. This does not rule out that, in addition to interlocus recombination, gene duplications occurred recently in stickleback. Duplications of MHC genes are more frequent in fish and bird species, as is also suggested by large interhaplotype differences in duplication number in several species (Malaga-Trillo et al. 1998; Hess and Edwards 2002).
Theoretical models predict that interlocus gene conversion can enhance the divergence between alleles (Ohta 1999). These expectations are consistent with the high mean nonsynonymous sequence polymorphism at peptide-binding residues up to dN ∼ 0.4 in our data. These values are in the range of the highest values for vertebrate populations, for example, the most polymorphic mammalian species, deer mouse (Richman et al. 2001). Interlocus recombination identified here among MHC class IIB loci may thus be a mechanism that is particularly important in light of the “divergent allele advantage,” i.e., when heterozygous individuals with more distantly related alleles have a fitness advantage over those with different but more similar alleles (Wakeland et al. 1990; Richman et al. 2003).
References
Bergström TF, Josefsson A, Erlich HA, Gyllenstein U (1998) Recent origin of HLA–DRB1 alleles and implication for human evolution. Nat Genet 18:237–242
Binz T, Reusch TBH, Wedekind C, Milinski M (2001) SSCP analysis of Mhc class IIB genes in the threespine stickleback. J Fish Biol 58:887–890
Bradley RD, Hillis DM (1997) Recombinant DNA sequences generated by PCR amplification. Mol Biol Evol 14:592–593
Britten RJ, Rowen L, Williams J, Cameron RA (2003) Majority of divergence between closely related DNA samples is due to indels. Proc Natl Acad Sci USA 100:4661–4665
Brown CJ, Garner EC, Dunker AK, Joyce P (2001) The power to detect recombination using the coalescent. Mol Biol Evol 18:1421–1424
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at the H-2 complex. Nature 256:50–52
Edwards SE, Wakeland EK, Potts WK (1995) Contrasting histories of avian and mammalian Mhc genes revealed by class II B sequences from songbirds. Proc Natl Acad Sci USA 92:12200–12204
Elsner HA, Rozas J, Blasczyk R (2002) The nature of introns 4–7 largely reflect the lineage specificity of HLA-A alleles. Immunogenetics 54:447–462
Fearnhead P, Donnelly P (2001) Estimating recombination rates from population genetic data. Genetics 159:1299–1318
Figueroa F, Mayer WE, Sültmann H, O’hUigin C, Tichy H, Satta Y, Takezaki N, Takahata N, Klein J (2000) Mhc class II B gene evolution in East African cichlid fish. Immunogenetics 51:556–575
Gu X, Nei M (1999) Locus specificity of polymorphic alleles and evolution by a birth-and-death process in mammalian MHC genes. Mol Biol Evol 16:147–156
Hess CM, Edwards SV (2002) The evolution of the major histocompatibility complex in birds. Biosience 52:423–431
Högstrand K, Böhme J (1994) A determination of the frequency of gene conversion in unmanipulated mouse sperm. Proc Natl Acad Sci USA 91:9921–9925
Hudson RR (2001) Two-locus sampling distributions and their application. Genetics 159:1805–1817
Hudson RR, Kaplan NL (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164
Hughes AL (1999) Adaptive evolution of genes and genomes. Oxford University Press, New York
Hughes AL (2000) Evolution of introns and exons of class II major histocompatibility comples genes of vertebrates. Immunogenetics 51:473–486
Hughes AL, Nei M (1988) Pattern of nucleotide substitution at major histocompatibility complex class I loci suggests overdominant selection. Nature 335:167–170
Klein J (1986) Natural history of the major histocompatibility complex. Wiley & Sons, New York
Kreitman M, Hudson RR (1991) Inferring the evolutionary histories of the adh and adh-dup loci in Drosophila melanogaster from patterns of polymorphism and divergence. Genetics 127:565–582
Kriener K, O’hUigin C, Tichy H, Klein J (2000) Convergent evolution of major histocompatibility complex molecules in humans and New World monkeys. Immunogenetics 51:169–178
Kumar S, Tamura K, Nei M (2004) MEGA version 3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163
Kupfermann H, Mayer WE, O’hUigin C, Klein D, Klein j (1992) Shared polymorphism between gorilla and human histocompatibility complex DRB loci. Hum Immunol 34:267–278
Langefors Å, Lohm J, Grahn M, Andersen Ø, von Schantz T (2001a) Association between major histocompatibility complex class IIB alleles and resistance to Aeromonas salmonicida in Atlantic salmon. Proc R Soc Lond Ser B 268:479–485
Langefors Å, Lohm J, von Schantz T (2001b) Allelic polymorphism in MHC class II B in four populations of Atlantic salmon (Salmo salar). Immunogenetics 53:329–336
Lee MSY (2001) Unalignable sequences and molecular evolution. Trends Ecol Evol 16:681–685
Malaga-Trillo E, Zaleska-Rutczynska Z, McAndrew B, Vincek V, Figueroa F, Sültmann H, Klein J (1998) Linkage relationships and haplotype polymorphism among cichlid Mhc class II B loci. Genetics 149:1527–1537
Martinsohn JT, Sousa AB, Guethlein LA, Howard JC (1999) The gene conversion hypothesis of MHC evolution: a review. Immunogenetics 50:168–200
McVean G, Awadalla P, Fearnhead P (2002) A coalescent-based approach for detecting and estimating recombination from gene sequences. Genetics 160:1231–1241
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nei M, Hughes AL (1992) Balanced polymorphism and evolution by the birth-and-death process in the MHC loci. In: Tsuji K, Aizawa M, Sasazuki T (eds) 11th Histocompatibility Workshop and Conference. Oxford University Press, New York, pp 27–38
Nei M, Li W-H (1980) Non-random association between electromorphs and inversion chromosomes in finite populations. Genet Res 35:65–83
Ohta T (1999) Effect of gene conversion on polymorphic patterns at major histocompatibility complex loci. Immunol Rev 167:319–325
Orti G, Bell MA, Reimchen TE, Meyer A (1994) Global survey of mitochondrial DNA sequences in the threespine stickleback: evidence for recent migrations. Evolution 48:608–622
Parham P, Ohta T (1996) Population biology of antigen presentation by MHC class I molecules. Science 272:67–74
Penn DJ, Damjanovich K, Potts WK (2002) MHC heterozygosity confers a selective advantage against multiple strain infections. Proc Natl Acad Sci USA 99:11260–11264
Posada D (2002) Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol Biol Evol 19:708–717
Potts WK, Manning CJ, Wakeland EK (1991) Mating patterns in seminatural populations of mice influenced by MHC genotype. Nature 352:619–621
Reusch TBH, Häberli MA, Aeschlimann PB, Milinski M (2001a) Female sticklebacks count alleles in a strategy of sexual selection explaining MHC polymorphism. Nature 414:300–302
Reusch TBH, Wegner KM, Kalbe M (2001b) Rapid genetic divergence in postglacial populations of threespine stickleback (Gasterosteus aculeatus): the role of habitat type, drainage, and geographical proximity. Mol Ecol 10:2435–2445
Reusch TBH, Schaschl H, Wegner KM (2004) Recent duplication and inter-locus gene conversion in major histocompatibility class II-genes in a teleost, the three-spined stickleback. Immunogenetics 56:427–437
Richman AD, Herrera LG, Nash D (2001) MHC class II beta sequence diversity in the deer mouse (Peromyscus maniculatus): implications for models of balancing selection. Mol Ecol 10:2765–2773
Richman AD, Herrera LG, Nash D, Schierup MH (2003) Relative roles of mutation and recombination in generating allelic polymorphism at an MHC class II locus in Peromyscus maniculatus. Genet Res Cambridge 82:89–99
Rozas J, Sanchez–DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP version 4: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 19:2496–2497
Sato A, Figueroa F, O’Huigin C, Steck N, Klein J (1998) Cloning of major histocompatibility complex (MHC) genes from threespine stickleback, Gasterosteus aculeatus. Mol Mar Biol Biotechnol 7:221–231
Sato A, Mayer WE, Tichy H, Grant PR, Grant BR, Klein J (2001) Evolution of Mhc class II B genes in Darwin’s finches and their closest relatives: birth of a new gene. Immunogenetics 53:792–801
Satta Y, O’Huigin C, Takahata N, Klein J (1993) The synonymous substitution rate of the major histocompatibility complex loci in primates. Proc Natl Acad Sci USA 90:7480–7484
Sawyer, SA (1999) Geneconv: a computer package for statistical detection of gene conversion. Code available at http://www.math.wustl.edu/∼sawyer
Stet RJM, Kruiswijk CP, Dixon B (2003) Major histocompatibility lineages and immune gene function in fish: the road not taken. Crit Rev Immunol 23:441–471
Strohbeck C (1983) Expected linkage disequilibrium for a neutral locus linked to a chromosomal arrangement. Genetics 103:545–555
Stumpf MPH, McVean GAT (2003). Estimating recombination rates from population-genetic data. Nat Rev Genet 4:959–968
Takahata N, Nei M. (1990) Allelic genealogy under frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124:967–978
Wakeland EK, Boehme S, She JX, Lu CC, McIndoe RA, Cheng I, Ye Y, Potts WK (1990) Ancestral polymorphism of MHC class II genes: divergent allele advantage. Immunol Res 9:115–122
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Pop Biol 7:256–276
Wegner KM, Kalbe M, Kurtz J, Reusch TBH, Milinski M (2003a) Parasite selection for immunogenetic optimality. Science 301:1343
Wegner KM, Reusch TBH, Kalbe M (2003b) Multiple parasite species are driving major histocompatibility complex polymorphism in the wild. J Evol Biol 16:224–232
Westerdahl H, Wittzell H, von Schantz T (2000) Mhc diversity in two passerine birds: no evidence for a minimal essential MHC. Immunogenetics 52:92–100
Wiehe T, Mountain J, Parham P, Slatkin M (2000) Distinguishing recombination and intragenic gene conversion by linkage disequilibrium patterns. Genetic Res 75:61–73
Yeager M, Hughes AL (1999) Evolution of the mammalian MHC: natural selection, recombination, and convergent evolution. Immunol Rev 167:45–58
Zangenberg G, Huang M-M, Arnheim N, Erlich H (1995) New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm. Nat Genet 10:407–414
Acknowledgments
We thank H. Schaschl for many valuable comments on the manuscript, S. Carstensen, S. Liedtke, N. Ryk, C. Schmuck, and T. Sonntag for laboratory assistance, and M. Milinski for ecouragement and support. Many thanks go to T. Reimchen for providing stickleback samples from British Columbia. TBHR thanks W. T. Stam for initially introducing him to indel alignment. TBHR was supported by Deutsche Forschungsgemeinschaft (DFG Re 1108/4 and -5). AL received a fellowship from the Swedish Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Richard Kliman]
Rights and permissions
About this article
Cite this article
Reusch, T.B., Langefors, Å. Inter- and Intralocus Recombination Drive MHC Class IIB Gene Diversification in a Teleost, the Three-Spined Stickleback Gasterosteus aculeatus. J Mol Evol 61, 531–541 (2005). https://doi.org/10.1007/s00239-004-0340-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0340-0