
Abstract
The interleukin-2 receptor (IL-2R) γ chain, or common γ chain (γc), is the hub of a protein interaction network in the mammalia that is central to defense against disease. It is the indispensable subunit of the functional receptor complexes for a group of interleukins known as the γ-chain-dependent interleukins (IL-2, IL-4, -7, -9, -15, and -21). The γc links these proteins through their interaction with it and their competition for its recruitment. The γc-dependent interleukins also interact with each other to either enhance or suppress expression through manipulation of expression of receptor subunits. Given the influence of protein–protein interactions on evolution, such as those documented for many genes including the reproductive proteins of the sperm and egg coat, here we have asked whether there is a common thread in the evolution of these interleukins. Our findings indicate that positive selection has acted by fixing a large number of amino acid replacement mutations in every single one of these interleukins, this adaptive evolution is also observed in a lineage-specific manner. Crucially, however, there does not appear to have ever been an instance of adaptive evolution in the γc chain itself, thereby providing an insight into the evolution of this hub protein. These findings highlight the importance of adaptive evolutionary events in the evolution of this central network in the immune system and suggest underlying causes for differences in defense responses in the mammalia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytokines typically function as intracellular messengers, mediating their effect via specific receptors on target cells. The principal functions of these cytokines include many complementary and many conflicting roles central to the induction, regulation, and functioning of the immune system in mammals (Kneitz et al. 1995; Lenardo 1991). Their function is governed by binding to a receptor bound to a T cell membrane. The receptor is responsible for transmitting a signal into the cell upon binding the appropriate ligand. This results in clonal expansion of T cells and, in some instances, stimulation of differentiation of T cells, B cells, NK cells, and myeloid cells (Leonard 1999). The γc-dependent cytokines bind to the receptors with varying degrees of affinity, based on the components of the receptor present (Leonard 1999). The interleukin-2 receptor (IL-2R), consisting of three subunits (αβγ), may be present in three forms: a low-affinity monomeric IL-2R form (α chain only), an intermediate-affinity dimeric IL-2R form (β and γ chains), and a high-affinity trimeric IL2R form (α, β, and γ chains). In order for IL-2 to bind to IL-2R there is a critical step that involves the binding of a high-mannose-type glycan, this carbohydrate binding site in IL-2 has been identified as Asn-26 (Fukushima et al. 2001). This formation induces cellular signaling and cell proliferation. The interleukins studied here share the signal transducing polypeptide IL-2Rγ or γc (Leonard 1999). The γc is also used by the IL-4, -7, -9, -15, and -21 receptors. Each of these cytokines also have a unique low-affinity receptor chain, the α chain, that is responsible for the specificity of the receptor to the particular cytokine. However, the γc chain implies that the transmembrane signal is similar for the different cytokine receptors (Leonard 1999).
There is well-documented evidence of important protein crosstalk between the γc chain-dependent cytokines, for instance, IL-7 selectively upregulates IL-2Rα and β chains (Chou et al. 1999), and IL-2 negatively regulates IL-7 receptor α-chain expression in activated T lymphocytes while upregulating the expression of IL-2Rα. (Xue et al. 2002). There are many similarities and interactions between these proteins at the level of receptor subunit recruitment and binding epitopes. The binding epitopes of γc-dependent cytokines are localized to four loops of the γc (Raskin et al. 1998). Mutational analysis of murine γc employing heterodimeric IL-2R and IL-7R on whole cells suggests that γc epitopes for IL-2 and IL-7 binding overlap and comprise at least three distinct putative loop segments of the γc protein (Olosz and Malek 2000). Binding epitope Tyr-103, for example, of the γc protein is a key ligand-interacting residue for IL-2, IL-4, and IL-7 and may be a common residue for all γc-dependent receptor systems (Zhang et al. 2002); others may overlap but need not be the same (He et al. 1995; Raskin et al. 1998). In addition, Tyr-103, Cys-161, Cys-210, and Gly-211, previously shown to contribute to binding IL-2 and IL-7, were also found to be involved in binding IL-4 and IL-15. Collectively, these data favor a model in which the γc chain utilizes a common mechanism for its interactions with multiple cytokines, and the binding sites are largely overlapping but not identical. Asn-128 and Tyr-103 likely act as contact residues, whereas Cys-161, Cys-210, and Gly-211 may stabilize the structure of the proposed ligand-interacting surface formed by the two extracytoplasmic domains (Olosz and Malek 2002).
Deletion or mutation of the γc chain of the receptor complex for these interleukins on the X chromosome results in severe immunological defects. Interestingly, the phenotypic effect of the XSCID disease differs between human and mouse (Mestas and Hughes 2004). In the human, it has been estimated that a number of mutations in the γc gene inhibit function of the immune response. The result in the majority of situations is a dramatic decrease in the number of T cells and NK cells, while B cell development is normal. However, in γc-null mice, B cell numbers are greatly diminished (Leonard et al. 1995). It has also been shown that IL-7R deficiency has different effects in both systems. In the mouse IL-7R deficiency blocks both T and B cell development (Peschon et al. 1994). IL-7R deficiency in the human only blocks T cell development (Roifman et al. 2000), leading to the suggestion that B cell development in the human is independent of IL-7 (Mestas and Hughes 2004). Clearly there has been a functional change related to IL-7. In this paper we have examined the role of natural selection in the evolution of this immunologically important group of signaling molecules.
The neutral and nearly-neutral theories of molecular evolution contend that the majority of sequence change is dictated by random genetic drift (Kimura 1983; Ohta 1992). If this is true, then positive selective pressures that have been capable of overcoming random genetic drift are likely to be very rare, but important. Adaptive evolution of colubine lysozyme genes permits foregut fermentation in these monkeys and is responsible for a functional shift in the protein, specifically an increased pH tolerance and resistance to digestion by pepsin (Messier and Stewart 1997; Stewart et al. 1987). Previously evolutionary differences have been noted at the sequence level between the human and the mouse (for a recent review see Emes et al. 2003); these include genes involved in olfaction (Gilad et al. 2003; Younger et al. 2001), reproduction (Torgerson et al. 2002), immunity, and host defense (Mestas and Hughes 2004). These positive selection events are usually correlated with functional shifts in those proteins in which we find these changes. Previous studies have shown that IL-2 is under positive selection (Zelus et al. 2000), however, we wished to determine in such a close network of interaction what kinds of selective pressures have influenced the evolution of these proteins.
Methods and Materials
We created alignments of five of the six known γc chain-dependent cytokine genes. IL-9 was omitted due to lack of sequence data. For each of these genes, the species, accession numbers and alignment lengths are listed in supplementary information Tables 1a and b. We have also created alignments of the available receptor sequences. These include the γc, the IL-2Rβ-chain receptor sequence, and the IL-2Rα-chain receptor sequence. We require a minimum of three sequences to allow directionality to our analyses with the use of an outgroup sequence that is more distantly related to the human and the mouse than they are to each other. These receptor sequences, species names, accession numbers, and alignment lengths are also available in the supplementary information. In all cases, there was at least one human and one mouse sequence. These sequences were available in both the GenBank and the Swissprot databases, thereby maximizing the likelihood of these sequences being correctly annotated and confirmed. All alignments were carried out at the amino acid level using the default settings in ClustalX 1.81 (Thompson et al. 1994), and gap positions were placed in the nucleotide sequences according to where they were found in the protein alignment. Phylogenetic trees for the data were constructed using the maximum likelihood criteria in the PAUP* program (Swofford 1998). An initial neighbor-joining phylogenetic tree was constructed and using this tree and maximum likelihood estimation, the transition–transversion ratio, rate matrix, base frequency, proportion of invariable sites, and shape of the rate distribution among sites were estimated. These values were then fixed and 1000 pseudosamples of the data were created using the bootstrap method (Felsenstein 1985). Phylogenetic hypotheses were constructed using these datasets and the results were summarized using a majority-rule consensus method. In all cases all internal branches were found in greater than 75% of all pseudosamples. Branch lengths are indicative of sequence divergence; see Figs. 1a–g for phylogenies.

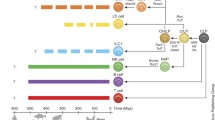
Phylogenetic representation of data used in analyses. a, b, c, d, e, f, and g are interleukin (IL)-2, IL-4, IL-7, IL-15, IL-21, IL-2 receptor subunit α, and the common γ chain, γc, respectively. The IL-2Rβ phylogeny is not shown, as it represents the relationship of three sequences and is trivial. Phylogenies were estimated using the maximum likelihood and all branches had high bootstrap support (>75%); these figures are omitted to avoid clutter. The phylogenies were unrooted for the analyses but are presented here rooted around the artiodactyl outgroup for clarity. The branch lengths correspond to the number of substitutions per site; the scale for each phylogeny is unique and is located below the phylogeny.
The likelihood ratio test (LRT) was used to evaluate a variety of models of codon sequence evolution (Yang 1998; Yang and Nielsen 2002; Yang et al. 2000). For all models used in this analysis a variety of starting ω values was taken and lnL scores were calculated for each. The starting ω value that generated the best lnL score was chosen, along with the corresponding parameter estimates. This was repeated a number of times to ensure that the same lnL score was reached. This process reduces the probability of using a local minimum on the likelihood plane rather than the global minimum. The LRT proceeds by comparing nested models of sequence evolution. These models usually allow for variable nonsynonymous-to-synonymous (dN/dS or ω) ratios among sites, along different branches of a phylogenetic tree, or in combination. These models imply that there are a variety of classes of sites in a given set of aligned sequences and the LRT provides a method of identifying the model that best describes the evolution of the set of sequences. One model is usually constrained so that ω ≤ 1. The more general model allows at least one class of sites to exist where the ω value is dependent on the data. In those cases where the ω value exceeds unity and the resulting increase in the likelihood score is significant, we can conclude that positive selection has occurred. In order to ascertain significance of difference in likelihood score we can compare the likelihood statistic 2Δl with χ 2v , where v is the number of degrees of freedom and corresponds with the number of free parameters.
The simplest model (fewest free parameters) is called M0. In this model it is assumed that there is a single ω value at all sites and across all lineages. This corresponds to the Goldman and Yang (1994) model. Model M1 assumes that there are two classes of sites—those with an ω value of 0 and those with an ω value of 1. Model M2 allows for three classes of sites—class 1 have an ω value of 0, class 2 an ω value of 1, and class 3 an ω value that is not fixed to any value and is estimated from the data. Given the relationship between M1 and M2, they can be tested for the significance of the difference of the fit of these two models using an LRT with df = 2. Model M3 allows all ω values to vary freely. There are two variants of this model employed in this analysis. The first is where there are two classes of sites that are free to vary (k = 2) and the second is where there are three classes of sites (k = 3). M3(k = 2) can be tested for its fit against M0 with df = 2. M3(k = 3) cannot be tested against any of the other models presented here using an LRT, however, its empirical comparison with M3(k = 2) can be interesting if the likelihood score is better.
We have also employed a number of models that use discrete approximations to continuous distributions in order to model variability in ω at different sites. The first of these, M7, assumes that variation in ω follows a beta distribution. A total of 10 classes of sites are assumed to exist and their ω values are constrained to be between 0 and 1. The second model, M8, allows the existence of another class of site where the ω value is allowed to be greater than unity. M8 and M7 can be compared with one another using the LRT with df = 2.
Finally, we used two models that allow the ω value to vary across sites and across different lineages. The first of these, MA, is a lineage-specific extension of M1 and the second, MB, is a lineage-specific extension of M3(k = 2). Both of these branch-site models can be compared with their site-specific counterparts using the LRT with df = 2. We identify those individual sites where positive selection is most likely to have occurred using the empirical Bayes approach as described previously (Nielsen and Yang 1998).
Results
To avoid repetition in this section, we will detail the likelihood ratio tests (LRTs) performed on the IL-2 sequence data specifically; see Table 1. All paml results are given in the supplementary information (Table 2–Table 8), and the summary is given in Table 2. The notation of the models is described under Materials and Methods. The LRTs performed include a χ2 test comparing: (1), model M0 with M3(k = 2), (2) M1 with M2, (3) M7 with M8, (4) M1 with model A, and (5) M3(k = 2) with model B, each with two degrees of freedom (df), and a comparison of M3(k = 2) with M3(k = 3). The same LRTs are performed for all other proteins in this analysis, and the resulting likelihood model calculated for each protein is discussed. Initial branch lengths were estimated from codon-based models.
The γc-dependent cytokines are composed of a common structural framework, the conformationally stable four-helix bundle, and rapid advances have been made in relating the structure and function of these cytokines (Chaiken and Williams 1996). There are currently 3-D structures available for IL-2 and IL-4, and a hypothetical structure for IL-7. Those sites identified in this study as being positively selected (greater than 95% posterior probability) are plotted on the available structures to give a structural and functional context for our findings.
IL-2 Analysis
The results for the analysis of IL-2 are given in Table 1. Our initial analysis involved using the original codon model described by Yang (Goldman and Yang 1994). For this model, denoted M0 in all tables, ω is constant for all sites and all positions. Under this model ω was estimated as 0.59207 (lnL = −2444.0929), indicating that, on average, the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site is less than unity. This likelihood score is one of the worst obtained for IL-2, indicating that it is a very poor fit to the data.
We then examined models of sequence evolution where the ω value is allowed to vary from site to site in the alignment. These models are indicated using the notation M1 to M8 for all proteins analyzed (see Tables 2–8 in supplementary data and Table 1 here for IL-2). Model M1 has two classes of sites, one class where ω is fixed at 0, implying that changes at these sites are unable to occur, and another class of sites where ω is fixed at unity. This model is a poor fit to the data (lnL = −2428.946626). Model M2 is an extension of M1 and allows an additional class of sites to exist, with ω estimated from the data. Under model M2, the likelihood score increases by 4 units (lnL = −2424.6353). This indicates that models that allow variation across the alignment provide a better description of the data than models that do not.
The next two models applied use an unconstrained discrete distribution in order to model variability at different sites in the alignment. The first of these models, M3(k = 2), allows two classes of sites to exist in the alignment. In order to determine whether a model which allows only one ω value over the entire alignment is sufficient to describe the evolution of the protein or whether a more complex model that allows for either two or three different categories of ω across the protein are necessary, models M3(k = 2) and M3(k = 3) are employed. For this reason a second LRT is performed to compare model M3(k = 2) to model M0 with df = 2. The LRT reveals that M3(k = 2) (lnL = −2424.6019) is significantly better than model M0, 2Δl = 38.91518, p < 0.0005, df = 2. It indicates that the first class of sites (representing 46% of the alignment) has an ω of 0.14224, indicating strong purifying selection, and the second class of sites (54% of the alignment) has an ω value that is only slightly greater than unity (1.04). This indicates a relaxed selective constraint on these sites, a small portion of which may be under positive selection. However, it is important to note that this is not the likelihood model for the data. Allowing three classes of sites M3(k = 3), the likelihood score obtained (lnL = −2424.2614) is not significantly better than the discrete model with two site classes. If there are sites under strong positive selection (as calculated by M3[k = 3]), they represent such a small proportion of the protein that any change in the resulting likelihood score is insignificant. We see a significant improvement in likelihood score with the inclusion of branch site-specific models.
The comparison of the site-specific models M7 and M8 indicate that there is no statistical improvement in the likelihood score when the LRT is performed, 2Δl = 1.25, with df = 2. This is not significant. However, model M7 indicates there may be a large proportion of sites (24%) that are selectively unconstrained; it is possible that a more suitable model will find that a small number of these may be positively selected.
The final category of models used are the branch-site models described by Yang (Yang and Nielsen 2002). These are designated “model A” and “model B” (Tables 2–8, supplementary information). Model A is an extension of the site-specific neutral model (M1) and can be compared with it using a χ2 test with df = 2. Treating the human and monkey branches individually as foreground (see Fig. 1a and Table 1), the difference in log likelihood is not significant (2Δl = 0.00094, df = 2, and 2Δl = 0.00078, df = 2, respectively). Treating the mouse branch as foreground, see Fig. 1a, the difference in log likelihood is highly significant (2Δl = 10.22994, df = 2, p < 0.005). This indicates that site- and lineage-specific models do provide us with a better description of the evolution of IL-2.
Model B is an extension of the unconstrained discrete distribution model M3(k = 2) and can be compared to it using a χ2 test with df = 2. We have labeled the human, monkey, and mouse branches in turn to determine if there has been lineage-specific evolution in any of these taxa. Treating the human and monkey branches independently as foreground, the LRT results indicate that model B is not significantly better than the discrete models (2Δl = 0.0008, df = 2, and 2Δl = 0, df = 2, for human and monkey branches, respectively), indicating that there are not lineage-specific evolutionary events detectable in these lineages. However, we find that the lowest likelihood score obtained for IL-2 is found using model B with the mouse branch treated as foreground. Performing an LRT between results for model B and results for the discrete model, M3(k = 2), we find that model B is statistically significantly better at the 1% significance level (2Δl = 9.8675, df = 2, p < 0.01). This model also indicates that some sites are evolving under the influence of positive selection, specifically in the mouse lineage. A total of 51% of the sites in the IL-2 data set are unconstrained in their evolution, with an ω value of 1.06, and 3% of sites are evolving under strong positive selection, ω = 176, in the mouse lineage only; see Table 2 for a summary of the likelihood model. The inference is that the IL-2 protein has been under increased positive selective pressure in the mouse lineage.
Using Bayesian estimations we have identified 35 sites for IL-2 that are under positive selection with greater than 0.95 posterior probability. These can be seen in Fig. 2a. The carbohydrate recognition site Asn-26 (volume of 114.1 Å3 and surface area of 160 Å2) is one of these positively selected sites. In the IL-2 alignment the Asn-26 site is highly conserved across all monkey species used and is also identical in the dog. In the rest of the phylogeny we observe variability at this site. In the rodent and artiodactyla, this amino acid residue is highly variable. In the mouse lineage the substitution at position 26 is for a serine, a tiny polar amino acid with a volume of 89.0 Å3 and a surface area of 115 Å2, therefore representing a radical substitution in the mouse lineage. In the pig and llama lineages, position 26 is a lysine, this is a positively charged nonpolar amino acid with a volume of 168.6 Å3 and a surface area of 200 Å2, overall quite similar in size and surface area to Asn. The other artiodactyl in the data set, the sheep, has a glutamate in position 26 which has a volume of 138.4 Å3 and a surface area of 190 Å2 and is a polar charged amino acid. The substitution in the rat and gerbil lineages is for an arginine, a positively charged polar residue with a volume of 173.4 Å3 and surface area of 225 Å2 that is physicochemically very similar to the lysine substitution in pig and llama. The most radical of all of these substitutions occurs in the mouse lineage, at a site that is known to be critical to the function of carbohydrate recognition.

The 3-D backbone structures of IL-2 (a, b) and IL-4 (c, e). For a and c, all sites evolving under negative or neutral evolution are dark colored and those under positive selection are light colored. For b, d, and e, sites involved in binding are light colored, while all other sites are dark colored. (a) IL-2 positively selected sites. (b) IL-2 receptor binding sites. (c) IL-4 positively selected sites; this view is rotated slightly to maximize the visualization of sites under positive selection. (d) IL-4 γc-chain binding epitopes. (e) IL-4 species-specific receptor-chain binding arms.
Previous work on the IL-2/IL-2R complex has identified a region 16 codons long responsible for binding to the receptor (Bubenik et al. 1987); these sites are highlighted in Fig. 2b. Of these 16 amino acid residues, 4 are identified here as having undergone positive selection. The binding sites of these interleukins interact with the γc chain by the first and fourth helical domains of IL-2 (Rose et al. 2003). Of the 35 positively selected amino acids, 8 are located in the first helix (A) and 2 are located in the last helix (D). This represents almost a third of all those positively selected sites. Earlier studies also suggested an interaction between these regions. Robb (1985) determined that amino acids at or near positions 8–27 and 33–54 are responsible for binding IL-2 to IL-2R. We have identified nine amino acids in this region as having undergone positive selection.
The same procedures and LRT tests were carried out for the remaining γc-dependent interleukins and the receptor sequences. What follows is a description of the maximum likelihood model that best fits the data for each interleukin and receptor chain analyzed; these are summarized in Table 2. (See Tables 2–8 supplementary information for all parameter estimates.)
IL-4 Analysis
The likelihood model for IL-4 is the discrete model with two site classes (M3[k = 2]) (see Table 2), the phylogeny used is described in Fig. 1b. Under this model, both proportions of sites and ω values are estimated from the data. It is estimated that 67% of the sites in the IL-4 protein are under purifying selection with an ω value of 0.1697 and 33% of sites are under positive selective pressure with an ω value of 1.16; see Table 2. Using Bayesian statistics there are 17 sites with posterior probabilities of being positively selected of greater than 0.95. The LRT test of M7 and M8 shows that M8 is a significantly better fit to the data (2Δl = 5.95, df = 2, p < 0.10). Model M8 finds 32% of the sites in the alignment are under positive selection (ω = 1.18) and also identifies a small proportion of these, 3%, or 15 sites, that have a high probability of having undergone strong positive selection (ω = 3.07).
The 3-D structure of IL-4 has been established (Walter et al. 1992). The 17 amino acids identified as having undergone adaptive evolution in IL-4 are highlighted in Fig. 2c. Of the 17 sites identified, 5 of these are located on either helix A or C. The binding epitope of IL-4 for IL-4Rα has been determined as a set of side chains determining the dissociation rate, Glu-9 and Arg-88 surrounded by five side chains (Ile-5, Thr-13, Arg-53, Asn-89, and Trp-91) of lower importance, and a partially overlapping set determining the association rate constant formed by five positively charged residues on helix C, Lys-77, Arg-81, Lys-84, Arg-85, and Arg-88 and two neighboring residues on helix A (Glu-9 and Thr-13). Of the positively selected sites, residue 12 is in close proximity to positions 9 and 13, both of which play an important role in α-chain receptor binding. Position 85 is under positive selection and forms part of this binding epitope for the α receptor chain; position 86 is also under positive selection. Position 89 is under positive selection; it forms part of the surrounding five side chains for binding to the IL-4 receptor and a neighboring amino acid 88, which is involved in binding to the α chain. There are a number of amino acid residues that are under positive selection and that flank a helix domain, for example, positions 32 and 34 mark the start and end of the B1 helix, position 58 marks the end of the B2 helix, and position 110 is the first amino acid of the D helix and all are under positive selection.
Residues important for receptor α-chain binding have been located on the antiparallel helices A and C (Kruse et al. 1993; Ramanathan et al. 1993). For mouse, it has been determined that the amino-terminal 16 residues and the carboxy-terminal 20 residues of IL-4 are required for species-specific receptor (α-chain) binding as well as T cell proliferation (Morrison and Leder 1992). These residues are highlighted on the IL-4 structure in Fig. 2e. A major proportion of the amino acid sequence between these regions can be substituted between human and mouse without any loss of receptor binding or biological function/activity (Morrison and Leder 1992). Glu-12, Ile-14, Leu-104, Asp-106, Phe-107, and Leu-111 bear side chains that are critical to IL-4 function. Glu-12 is under positive selection in our data set, as is position 110. A small number of sites responsible for the binding of IL-4 to the γc chain have been determined to date (Zhang et al. 2002) (see Fig. 2d), one of which (position 128) is within a distance of three amino acid residues of a positively selected amino acid.
IL-7 Analysis
For IL-7 the best model was the site-specific discrete model M3(K = 2); see Table 2 for a summary of the likelihood model and Fig. 1c for the phylogeny used. The results of this model are also supported by the LRT test between M7 and M8 (2Δl = 6.45, df = 2, p < 0.05). Model M8 is a significantly better fit to the IL-7 data than M7, and it predicts that ∼14% of the protein is under positive selection with an ω value of 3.07. Model M8 also identifies two amino acid positions under positive selection, these positions are 71 and 79 and are both located in the C helix of the protein. Under the M3(k = 2) model, it is estimated that there are 81% of the sites under strong purifying selection, with ω estimated as 0.2928, and 19% of sites under positive selection, with an ω estimated as 2.6. Using Bayesian statistics, 26 sites are classified as having a >0.5 posterior probability of belonging to the positively selected class of sites across all lineages, and 7 of these sites (including those identified using model M8) are classified as having a >0.95 posterior probability. As only seven sites in our analysis had >0.95 posterior probability, we do not include the 3D structure here, but supply a brief summary of their location. The sites in question are located only on the helices of the IL-7 protein, three sites in helix A, two sites in helix B, one site within helix C, and one site in helix D. The helices are known to be important for receptor binding from studies of other members of this group of cytokines.
IL-15 Analysis
For IL-15, the phylogeny used is shown in Fig. 1d and the likelihood model is the site-specific discrete model with three site classes, M3(k = 3). Under this model (Table 2), there is a large portion (70%) of the alignment under strong purifying selection (ω = 0.1259), 29% of sites have a relaxed selective constraint with an ω value of 1.13 and the final class of sites estimated from the data has an ω value of 14.82. However, this is a tiny proportion of sites (1%). It is estimated from Bayesian statistics that 38 specific sites have a >0.5 posterior probability of belonging to the category of amino acids with an ω value of 1.13, and of these, 11 have >0.95 posterior probability of being in the positively selected category with an ω value of 14.82. The LRT test of M7 versus M8 reveals that M8 is a better fit to the IL-15 data at the 10% significance level (2Δl = 5.54, df = 2, p < 0.10), but not at the 5% level, which has a critical value of 5.99. Model M8 indicates that a small proportion (1%) of the protein is under strong positive selection ω = 13.48. Very few data exist concerning the residues on IL-15 involved in the binding of the various receptor subunits. However, it is thought that binding sites for IL-15 and IL-2 may be similar for IL-2Rβ and γc subunit binding (Pettit et al. 1997).
IL-21 Analysis
The maximum likelihood model for IL-21, is M3(k = 2), the phylogeny used is described in Fig. 1e. Under this model (Table 2), it is estimated that 77% of sites in the IL-21 protein are under purifying selection with an ω value of 0.22944, and 23% of sites under a positive selection with an ω value of 1.25. From Bayesian statistics it is estimated that 20 sites have >0.5 posterior probability of being under positive selection, 2 of these sites with strong support (0.95 posterior probability). However, the LRT test of M7 (lnL = −1223.225) versus M8 (lnL = −1222.949) does not describe a statistically significant increase in lnL for M8. M8 estimates that a large amount of the protein sequence, ∼23%, is under positive selective pressure (ω = 1.25); a slight increase in lnL is apparent.
Sufficient data exist for the γc chain and the IL-2Rα and β chains; the phylogenies used for IL-2Rα and γc are shown in Figs. 1f and g, respectively. As there are only three sequences for IL-2Rβ, the phylogeny is trivial and is not shown.
IL-2Rα Analysis
For IL-2Rα, the best model tested for site specific change was the discrete model with three site classes (M3[k = 3]), as outlined in Table 2. Under this model there are three categories of sites with ω values estimated from the data. Strong purifying selection is detected in 39% of the sites in the α chain with an ω value of 0.0948, 56% of the sites are under weak negative selection with an ω value of 0.77758, and 5% of the sites are under positive selection for change with an ω value of 1.94. There are six sites selected using Bayesian statistics as having >0.5 posterior probability of being under positive selection; these are at positions 20, 66, 68, 177, 178, and 200 in the amino acid sequence. Position 20 is at the end of the signal domain of IL-2Rα, positions 66 and 68 are just before the carbohydrate binding position, which is amino acid 70, positions 177 and 178 are part of the second Sushi domain, and position 200 is potentially part of the extracellular domain. The Bayesian support for these sites is very low and so must be treated with caution. Although not statistically significant, there is an increase in lnL when M8 is compared to M7. Perhaps because the number of amino acids (4% of sites) showing evidence of positive selection (ω = 1.94) does not comprise a large enough portion of the overall alignment that any addition of parameters is not justified mathematically, M7 is chosen as the more likely model for the data. M7 describes the β distribution of of ω values among sites as having a large portion of amino acids with ω values close to 0, a large portion of the protein with ω values close to unity, and the remainder distributed evenly between the two extremes.
IL2Rβ Analysis
The results for IL2Rβ also indicate positive selection at work on this chain of the receptor complex (Table 2). The best model following LRT calculations M3(k = 3). The LRT test between M7 and M8 also indicates positive selection (2Δl = 13.426, df = 2, p < 0.001). Under model M8, 2% of the protein has signatures of positive selection. Under model M3(k = 3), 61% of the sites are estimated to have an ω value of 0.12264 and are under strong negative selection, 37% of the sites have an ω value of 1.18, and 2% of the sites (11 sites) are under strong positive selection with an ω estimated at 999. These 11 sites include positions 4, 105, 342, 345, 388, 455, 515, 517, 520, 522, and 528. Position 4 is part of the signal sequence and position 106 marks the end of a strand region of the protein. The remaining nine amino acid positions all fall within the cytoplasmic domain of the protein and are in close succession on the protein sequence. It is possible that the large proportion of sites (37% of the protein) with ω values slightly greater than 1 represents a cohort of sites in the IL2Rβ protein that allow for a compensatory effect against those radical or diversifying substitutions that are positively selected. In total, there are 42 sites identified as positively selected with a posterior probability of 0.95 using Bayesian statistics. Of these 42 sites, 2 are part of the signal domain for IL2Rβ, 27 are part of the cytoplasmic domain, 5 are part of the fibronectin type-III domain, position 31 is only 2 positions from the carbohydrate binding site and is under positive selection, 5 sites are at the start of strand domains in the secondary structure of this protein, 1 site is at the end of a turn, 4 sites are in the spaces between strands, and 1 site is between a strand and a helix. Position 266 is under positive selection and is the first amino acid position after the transmembrane domain, and position 219 is the last site before the WSXWS motif. The only domain of the IL2Rβ protein without any positively selected site present is the transmembrane domain. The lack of positive selection in the transmembrane domain perhaps indicates the chemical constraint on amino acids that span the membrane and highlights the importance of the signal transducing domain. Alternatively this region could have no neutrally evolving sites.
γc Analysis
The best model tested for the γc chain is the discrete model with two site classes (M3[k = 2]), as outlined in Table 2. Under this model it was estimated that 38% of sites are under very strong purifying selection with an ω estimated at 0.00001, and the remaining 62% of sites are under slightly weaker purifying selection, with an ω value estimated at 0.56591. These results indicate that any change in the γc chain of the receptor is selected against. The LRT test between M7 (lnL = −3185.508) and M8 (lnL = −3185.095) shows M7 to be the better model. M7 also supports this strong purifying selection, describing a β distribution with the majority of sites in the protein with very low ω values (<0.25).
The results show that of all the known γc-dependent cytokines (with the exception of IL-9 which will involve further sequencing before analysis of this kind can be carried out) and receptor sequences, all show evidence of positive selection, either in a branch site- or in a site-specific manner with the one exception of the hub protein, γc, which is under strong purifying selection in all mammalian lineages tested and across all sites.
Discussion and Conclusion
We have identified heterogeneous selective pressures, including positive selection, relaxation of selective constraints, and purifying selection in the γc-dependent cytokines and the receptor chains tested. However, for the γc there is evidence of strong purifying selection. The overwhelming majority of positively selected sites in the ligands occurs in those regions known to be involved in binding to the receptor complex. We therefore believe that the selective pressure at work in this network involves a more efficient and competitive binding and attraction of receptor chains by the cytokines. As the γc is the central protein in this protein interaction network, it is under a strong selective pressure to remain unchanged. All other proteins in the network compete for the recruitment of the γc and we suggest that, in doing so, have become subject to positive selection for change. We have found IL-2 to exhibit a relaxed selective constraint across all of the mammalian species tested, with a total of 51% of the sites with an ω value of 1.08. There is a small proportion of sites (3%) in IL-2 under positive selection in the mouse lineage alone with an ω value of 163. We have identified 22 sites belonging to the positively selected category, one of which (Asn-26) is the carbohydrate binding site of IL-2 which is responsible for the initial binding of IL-2 to a high mannose-type glycan; this is the first step in the binding of IL-2 to the receptor. The Asn-26 position is present in all primate species and in dog, but this position is highly variable among the rest of the phylogeny, with the most radical of all the substitutions at this position occurring in the mouse lineage (Asn to Glu).
A significant proportion of all those sites identified as being under positive selection in IL-2 is located in regions of the protein involved in receptor binding. It is of particular interest in this light that a statistically significantly large portion of these sites found to be under positive selection is located in those regions important for binding to the β and γc chains of the receptor. Of the 35 sites identified using Bayesian statistics as belonging to the positively selected category, 8 are located in helix A of IL-2 including the previously mentioned carbohydrate binding site, Asn-26. The A helix of IL-2 is known to play a role in the binding of the γc and β chains. In total, 25 amino acids of 35 under positive selection are located on helices, the majority of which are on helices A, B, and C. There are also a number of positively selected amino acids occurring at regions flanking domains; these sites may have a central role in defining a new conformation.
Il-2Rα and β exist in part as preformed complexes in which the affinity of IL-2Rβ for IL-2 is altered by the proximity of IL-2Rα through mechanisms that do not require the prior binding of IL-2 to IL-2Rα (Roessler et al. 1994). Both of these subunits effect IL-2 regulation and affinity by interacting and binding to each other, and we have identified positive selection in both of these. There is evidence that ligand-dependent receptor internalization may be the rate-limiting step controlling the duration of IL-2R signaling; a reduction in the receptor internalization rate could contribute to the observed response enhancement (Chang et al. 1996). Previous analysis of the IL-2Rβ chain has revealed a region between the two Stat5 docking sites in the IL-2Rβ chain that mediates signaling events that, together with Stat5 activation, are essential for the stimulation of IL-2Rα gene transcription (Imbert et al. 2002). This region is between position 392 and position 510 of the IL2Rβ chain; we have found 10 positively selected amino acids (∼10%) in this region. Perhaps, a more reduced rate of internalization and therefore more enhanced IL-2R signaling as well as more efficient IL-2Rα transcription have resulted from the functional shift produced by positive selection in these proteins. Equally important, the IL-2 protein itself has undergone positive selection in the regions responsible for receptor binding, demonstrating a complementary effect in the evolution of ligand and receptor.
It is perhaps of no surprise to find positive selection in IL-4, as it has already been observed that positive selection drives the regulation of this gene in human populations (Rockman et al. 2003). Positive selection on the IL-4 promoter has had the effect of altering the whole balance of interactions within the immune system by eliciting a threefold higher expression in the human lineage, and now we have found that the protein itself is under positive selection. This protein is known to play a central role in viral infection. For IL-4, of the 16 amino acids identified as belonging to the positively selected category, 5 are located on either the A or the C helix. These helices are involved in binding to the receptor. A number of individual amino acids directly involved in binding to the α-chain receptor are under positive selection. In those cases where we have analyzed the results in a structural framework, there is a large proportion of sites under positive selection that are at the start or end of domains such as helices or strands. These regions are conformationally important and may play a role in altering function by changing structure; it has previously been shown that a single amino acid substitution can alter the antigen-binding specificity of a protein (Rudikoff et al. 1982).
The results from the IL-7 analysis are interesting in light of the known differences in functionality of this protein in the human and mouse species. Positive selection is supported by a number of models across the mammalian phylogeny, and the sites under positive selection directly correspond to regions believed to be involved in receptor binding. It is possible that the functional shift predicted to have occurred in these specific regions could have contributed to the development of an IL-7-independent B cell development in the human lineage. To date, very few data exist for residues in the IL-15 and IL-21 proteins that are involved in receptor binding, but preliminary studies suggest the regions in which we have found positive selection correspond to regions involved in receptor binding.
Analysis of the IL-2Rα, -β, and -γc of the receptor complex shows that both the α and the β chains are under positive selection and the γc is under negative selection. This perhaps makes sense as the γc is involved in so many other interactions that a change in this one protein would have a huge effect in the overall protein interaction pathway (Goh and Cohen 2002). It was found that in the IL2Rα protein, all the positively selected sites are located in regions other than the transmembrane domain, including sites in extremely close proximity to the carbohydrate binding position. In the IL-2Rβ analysis sites again very close to the carbohydrate binding position were identified as being under positive selection; the only domain of this protein that does not show evidence of positive selection is the transmembrane domain. This may be due to the constraint placed on this region of the protein due to its importance in anchoring of the receptor subunits. We have found 10% of the sites in the gene transcription regulatory region of IL2Rβ to be under positive selection. This is likely to have a significant biological impact.
The results presented here suggest a general positive selective pressure acting on a whole network of interacting proteins. The only protein in the network that is under strong purifying selection is the protein at the hub of this network, thus showing that selective pressure on the central protein in the network is greater than that of the interacting and competing proteins. This may be driving the functional shifts within the cell-to-cell communicators of the mammalian immune system. We believe the findings in this study show that the mechanism driving the evolution of these cytokines and receptors is the competition for recruitment of the γc and is strongly related to ligand–receptor binding and affinity.
References
Bubenik J, Krchnak V, Indrova M, Hamsikova E, Jandlova T, Simova J, Kieler J (1987) Localization of a receptor binding site on the IL-2 molecule. Folia Biol (Praha) 33:266–281
Chaiken IM, Williams WV (1996) Identifying structure–function relationships in four-helix bundle cytokines: towards de novo mimetics design. Trends Biotechnol 14:369–375
Chang DZ, Wu Z, Ciardelli TL (1996) A point mutation in interleukin–2 that alters ligand internalization. J Biol Chem 271:13349–3355
Chou YK, Bourdette DN, Barnes D, Finn TP, Murray S, Unsicker L, Robey I, Whitham RH, Buenafe AC, Allegretta M, Offner H, Vandenbark AA (1999) IL-7 enhances Ag-specific human T cell response by increasing expression of IL-2R alpha and gamma chains. J Neuroimmunol 96:101–111
Emes RD, Goodstadt L, Winter EE, Ponting CP (2003) Comparison of the genomes of human and mouse lays the foundation of genome zoology. Hum Mol Genet 12:701–709
Felsenstein J (1985) Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791
Fukushima K, Hara-Kuge S, Ideo H, Yamashita K (2001) Carbohydrate recognition site of interleukin-2 in relation to cell proliferation. J Biol Chem 276:31202–31208
Gilad Y, Man O, Paabo S, Lancet D (2003) Human specific loss of olfactory recpetor genes. Proc Natl Acad Sci USA 100:3324–3327
Goh CS, Cohen FE (2002) Co–evolutionary analysis reveals insights into protein–protein interactions. J Mol Biol 324:177–192
Goldman N, Yang Z (1994) A codon–based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725
He YW, Adkins B, Furse RK, Malek TR (1995) Expression and function of the gamma c subunit of the IL-2, IL-4, and IL-7 receptors. Distinct interaction of gamma c in the IL–4 receptor. J Immunol 154:1596–1605
Imbert V, Rezzonico R, Reichenbach P, Nabholz M (2002) Induction of interleukin-2 receptor alpha (IL–2Ralpha) expression by interleukin-2: important role of the interleukin-2 receptor beta chain region between the two Stat5 docking sites. Eur Cytokine Network 13:331–339
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge
Kneitz B, Herrmann T, Yonehara S, Schimpl A (1995) Normal clonal expansion but impaired Fas-mediated cell death and anergy induction in interleukin-2-deficient mice. Eur J Immunol 25:2572–2577
Kruse N, Shen BJ, Arnold S, Tony HP, Muller T, Sebald W (1993) Two distinct functional sites of human interleukin 4 are identified by variants impaired in either receptor binding or receptor activation. EMBO J 12:5121–5129
Lenardo MJ (1991) Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 353:858–861
Leonard WJ (1999) Fundamental immunology. Lippincott-Raven, Philadelphia
Leonard WJ, Shores EW, Love PE (1995) Role of the common cytokine receptor gamma chain in cytokine signaling and lymphoid development. Immunol Rev 148:97–114
Messier W, Stewart CB (1997) Episodic adaptive evolution of primate lysozymes. Nature 385:151–154
Mestas J, Hughes CCW (2004) Of mice and not men: Differences between mouse and human immunology. J Immunol 172:2731–2738
Morrison BW, Leder P (1992) A receptor binding domain of mouse interleukin-4 defined by a solid-phase binding assay and in vitro mutagenesis. J Biol Chem 267:11957–11963
Nielsen R, Yang ZH (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Ohta T, (1992) The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst 23:263–286
Olosz F, Malek TR (2000) Three loops of the common gamma chain ectodomain required for the binding of interleukin-2 and interleukin-7. J Biol Chem 275:30100–30105
Olosz F, Malek TR (2002) Structural basis for binding multiple ligands by the common cytokine receptor gamma-chain. J Biol Chem 277:12047–12052
Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL (1994) Early lymphocyte expansion is severely impaired in interleukin-7 receptor-deficient mice. 180:1955–1960
Pettit DK, Bonnert TP, Eisenman J, Srinivasan S, Paxton R, Beers C, Lynch D, Miller B, Yost J, Grabstein KH, Gombotz WR (1997) Structure–function studies of interleukin 15 using site-specific mutagenesis, polyethylene glycol conjugation, and homology modeling. J Biol Chem 272:2312–2318
Ramanathan L, Ingram R, Sullivan L, Greenberg R, Reim R, Trotta PP, Le HV (1993) Immunochemical mapping of domains in human interleukin 4 recognized by neutralizing monoclonal antibodies. Biochemistry 32:3549–3556
Raskin N, Jakubowski A, Sizing ID, Olson DL, Kalled SL, Hession CA, Benjamin CD, Baker DP, Burkly LC (1998) Molecular mapping with functional antibodies localizes critical sites on the human IL receptor common gamma (gamma c) chain. J Immunol 161:3474–3483
Robb RJ (1985) Interleukin-2 and its cell-surface receptor. Behring Inst Mitt 77:56–67
Rockman MV, Hahn MW, Soranzo N, Goldstein DB, Wray GA (2003) Positive selection on a human-specific transcription factor binding site regulating IL4 expression. Curr Opin Biol 13:2118–2123
Roessler E, Grant A, Ju G, Tsudo M, Sugamura K, Waldmann TA (1994) Cooperative interactions between the interleukin 2 receptor alpha and beta chains alter the interleukin 2-binding affinity of the receptor subunits. Proc Natl Acad Sci USA 91:3344–3347
Roifman CM, Zhang JY, Chitayat D, Sharfe N (2000) A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Proc Natl Acad Sci USA 96:2803–2807
Rose T, Moreau JL, Eckenberg R, Theze J (2003) Structural analysis and modeling of a synthetic interleukin-2 mimetic and its interleukin-2Rbeta2 receptor. J Biol Chem 278:22868–22876
Rudikoff S, Giusti AM, Cook WD, Scharff MD (1982) Single amino acid substitution altering antigen-binding specificity. Proc Natl Acad Sci USA 79:1979–1983
Stewart CB, Schilling JW, Wilson AC (1987) Adaptive evolution in the stomach lysozymes of foregut fermenters. Nature 330:401–404
Swofford DL (1998) PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates, Sunderland, MA
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Torgerson DG, Kulathinal RJ, Singh RS (2002) Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol Biol Evol 19:1973–1980
Walter MR, Cook WJ, Zhao BG, Cameron RPJ, Ealick SE, Walter RLJ, Reichert P, Nagabhushan TL, Trotta PP, Bugg CE (1992) Crystal structure of recombinant human interleukin-4. J Biol Chem 267:20371–20376
Xue HH, Kovanen PE, Pise–Masison CA, Berg M, Radovich MF, Brady JN, Leonard WJ (2002) IL-2 negatively regulates IL-7 receptor alpha chain expression in activated T lymphocytes. Proc Natl Acad Sci USA 99:13759–13764
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15:568–573
Yang Z, Nielsen R (2002) Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol 19:908–917
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Younger RM, Amadou C, Bethel G, Ehlers A, Fischer Lindahl K, Forbes S, Horton R, Milne S, Mungall A, Trowsdale J, Volz A, Ziegler A, Beck S (2001) Characterization of clustered mhc-linked olfactory receptor genes in human and mouse. Genome Res 11:519–530
Zelus D, Robinson-Rechavi M, Delacre M, Auriault C, Laudet V (2000) Fast evolution of interleukin-2 in mammals and positive selection in ruminants. J Mol Evol 51:234–244
Zhang JL, Buehner M, Sebald W (2002) Functional epitope of common gamma chain for interleukin-4 binding. Eur J Biochem 269:1490–1499
Acknowledgments
We would like to thank the HEA for funding this work under the PRTLI scheme. We would also like to thank Dr. Christopher J. Creevey and Dr. Simon A. Travers for their contribution to the phylogeny reconstruction.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Willie J. Swanson]
Rights and permissions
About this article
Cite this article
O’Connell, M.J., McInerney, J.O. Gamma Chain Receptor Interleukins: Evidence for Positive Selection Driving the Evolution of Cell-to-Cell Communicators in the Mammalian Immune System. J Mol Evol 61, 608–619 (2005). https://doi.org/10.1007/s00239-004-0313-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0313-3