
Abstract
Thirty-three percent (228/682) of all long terminal repeat (LTR) retrotransposon sequences (LRSs) present in the sequenced Drosophila melanogaster genome were found to be located in or within 1000 bp of a gene. Recently inserted LTR retrotransposons are significantly more likely to be located in or within genes than are older, fragmented LTR retrotransposon sequences, indicating that most LRS-gene associations are selected against over evolutionary time. LRSs associated with conserved genes (homologenes) are especially prone to negative selection. In contrast, fragmented LRSs that have persisted in the genome over long spans of evolutionary time are preferentially associated with genes involved in signal transduction and other newly evolved functions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) are mobile sequences abundant within eukaryotic genomes (e.g., Drosophila melanogaster, 10–20% [Hoskins et al. 2002; Kaminker et al. 2002); Homo sapiens, >40% [Li et al. 2001]; Lillium, >90% [Leeton and Smyth 1993]). Although TEs may be maintained in populations on a day-to-day basis even in the face of slight negative selection (e.g., Charlesworth et al. 1997; Doolittle and Sapienza 1980; Hickey 1982; Orgel and Crick 1980), this does not preclude the possibility that TE sequences may contribute significantly to gene and genome evolution over evolutionary time (e.g., McDonald 1993, 1995). Indeed, there are now many examples of TE sequences having contributed significantly to gene and genome evolution in a variety of species (e.g., Brosius 1999; Makalowski 2000; Medstrand et al. 2001). With the availability of sequence databases for a number of species, it has become possible to conduct systematic genome searches for TE-gene associations in order to objectively assess the potential contribution of these elements to gene evolution. For example, recent analyses in the human genome have shown that retrotransposon sequences are present in the protein coding regions of ∼4% of genes (Nekrutenko and Li 2001), in the untranslated regions (UTR) of ∼27% of genes (van de Lagemaat et al. 2003), and in ∼25% of promoter regions (Jordan et al. 2003). We recently reported that long terminal repeat (LTR) retrotransposon sequences are present within the regulatory region and/or the transcription boundaries of 0.6% of C. elegans genes (Ganko et al. 2003). In this paper, we report the results of a detailed analysis of the association of LTR retrotransposon element sequences (LRSs) with genes in the sequenced Drosophila melanogaster genome. (The term LRSs here refers to all full-length LTR retrotransposons, all solo LTRs, and any fragmented derivatives thereof.)
LTR retrotransposons are a class of transposable elements that have a life cycle analogous to that of infectious retroviruses (Boeke et al. 1985). LTR retrotransposons are initially transcribed into RNA by the host organism’s transcriptional machinery and subsequently reverse transcribed by element-encoded reverse transcriptase (RT) to create a DNA copy. In order to initiate RNA transcription, LTR retrotransposons contain cis-regulatory sequences typical of eukaryotic genes, including promoter, enhancer, and termination signals. The regulatory effects of these signals are not limited to the LRS in which they are contained, and may influence the expression of adjacent genes. In addition, LRSs may also be incorporated into the coding regions of genes. Thus, LRS insertions that do not destroy gene function may be a potential source of adaptive genetic variation (e.g., Brosius 1999; Makalowski 2000; McDonald 1993, 1995).
Drosophila melanogaster is good model for evolutionary genomic studies because of the availability of a high-quality genome sequence (Adams et al. 2000; Celniker et al. 2002) and annotation (Misra et al. 2002), especially with regard to TEs (Kaminker et al. 2002). We report here the identification and preliminary characterization of 82 LRSs located within 1 kb of a gene and an additional 146 LRSs located inside gene boundaries. Genes with LRSs located within their boundaries are significantly larger (∼5×) than the average D. melanogaster gene. LRSs are preferentially associated with recently evolved genes encoding signal transduction functions.
Methods
LRS-Gene Association Data
Annotated chromosome files (Release 3.1) were downloaded from the Berkeley Drosophila Genome Project Web site (ftp://ftp.fruitfly.org/pub/download/dmel_RELEASE3-1/FASTA/) in Spring 2003. The distance from each annotated LRS (Kaminker et al. 2002) to the closest flanking gene on each side of the LRS was determined, with the exception of the centromere and telomere termini where a transposon may have only one flanking gene. We filtered these results by defining an LRS-gene association as an LRS ≤1000 bp of a gene, based on reports that most D. melanogaster cis-regulatory sequences lie within 1 kb of transcriptional boundaries (Papatsenko et al. 2002). Thus, all LRSs included in our analyses were either in gene boundaries or within 1000 bp of a gene (Table 1). We define “internal association” as an LRS inside the defined transcription borders of a gene and define “proximal association” as an LRS within 1–1000 bp of gene boundaries. Expectation values for associations in Table 1 were determined using the distribution ratio of each LRS family; that is, the number of individuals in a given LRS family divided by the sum of all identified LRSs. This family distribution value was multiplied by the sum of all associations, the sum of internal LRS-gene associations, or the sum of proximal associations to provide an expectation for a given LRS family in the respective category.
Size and Density Analyses
Information regarding the function, size, chromosomal position, gene ontology, and expression of each gene was collected primarily from Flybase (http://flybase.bio.indiana.edu/) gene reports (Spring 2003 data releases). Gene size was determined using the most distant start and stop nucleotides in the case of multiple transcripts. Homologous gene data were obtained from the Homologene database (http://www.ncbi.nlm.nih.gov/HomoloGene/), an NCBI- generated dataset of putative orthologous genes between important model organisms (Wheeler et al. 2003). Orthology between genes is generally believed to imply functional conservation. Tests of a distribution model for LRS internal and proximal to genes were carried out by binomial tests as described previously (Ganko et al. 2003).
To measure gene density, each chromosome was divided into successive 200-kb regions, and the number of genes in each region summed. The gene density of each bin was calculated for the entire chromosome, then for all regions that contained at least one LRS, and, finally, for regions that contained an LRS-gene association. As a second measure of gene density, we compared the mean intergenic distance between all genes to the intergenic distance of genes with an associated LRS.
Consensus sizes for individual families were determined from Flybase (Kaminker et al. 2002) or RepBase (Jurka 2000), and the size of each individual LRS element was compared to the size of the consensus sequence for the appropriate LRS family to calculate the “percentage consensus size.” The results were separated into three categories: near-full-length (LRS ≥90% of the consensus size), medium (21–89% of consensus size), and small (LRS ≤20% of consensus size). Expectation values were calculated based on the ratio of LRSs in a given size bin to all LRSs in the genome.
Functional Analysis of Genes
Genes were classified into functional categories based on Gene Ontology (GO) terms. The Gene Ontology project has created a controlled vocabulary describing the functional products of genes (Ashburner et al. 2000; Harris et al. 2004). To investigate this defined hierarchical classification we created a set of Perl scripts (Greene, Ganko, and McDonald, in preparation) to trace genes from a specific GO ID to the general descriptors. For example, the ID GO0004871 has a specific description of “signal transducer activity” as a general “molecular function.” For every descriptor GO ID there exist one or more paths to the most general root terms (‘cellular component,’ ‘biological process,’ ‘molecular function’). In the case of multiple paths, we trace each GO ID through all possible routes. Performing a trace on a set of genes results in a functional profile that can then be compared to the functional profile of other gene sets. Chi-square tests were used for initial profile comparisons, followed by binomial tests on individual descriptor terms. We used a Bonferroni correction as an adjustment for multiple comparisons in all binomial p-values.
Results
One-Third of All Identified LRSs in the Sequenced D. melanogaster Genome Are Located in or Within 1000 bp of a Gene
The D. melanogaster genome is estimated to contain approximately 13,300 genes (Adams et al. 2000; Misra et al. 2002). Recently, 682 LRSs have been identified in the euchromatic portion of the Drosophila genome (Kaminker et al. 2002). We developed Perl scripts to determine the distance from each of these 682 LRSs to the nearest flanking genes. Since most sequences known to exert cis-regulatory effects on Drosophila gene expression are located within 1000 bp of the transcriptional start site (Papatsenko et al. 2002), we limited our dataset to genes with LRS located within 1000 bp upstream or downstream of established genes or within gene boundaries (introns or exons). This dataset contains LRS-gene associations of potential adaptive significance.
Our results (Table 1) indicate that 228 (33.4%) LRSs located in the euchromatic region of the Drosophila genome are associated with genes. There are 82 (12.0%) LRS sequences located 1 kb upstream or downstream of 102 genes (proximal associations). (Note that because some LRSs are located within 1 kb of two genes, the number of associated genes is greater than the number of associated LRS.) There are an additional 121 (21.4%) LRSs located within the introns of genes and 25 LRSs located in both introns and exons of genes, for a total of 146 internal associations. Proximal associations are comprised of element sequences distributed equally upstream and downstream of genes (53/102 upstream, 49/102 downstream). Likewise, there is no significant bias in the sense orientation of element sequences located proximal to genes (upstream, 30/53 elements in sense orientation with respect to the associated gene; downstream, 23/49 in sense orientation; χ2 p > 0.10). Nor did we identify a significant bias toward antisense orientation (63/146 in sense orientation; p > 0.09) when looking at LRS associations inside genes. Our result contrasts with a recent study of the human genome where it was found that LRSs located in human genes are most often in an antisense configuration with respect to the gene (Medstrand et al. 2002), while retrotransposons in the 5′ and 3′ untranslated regions are significantly more likely to be in a sense configuration (van de Lagemaat et al. 2003). However, our results in Drosophila are similar to the relatively equal sense/antisense orientation of LRSs located proximal to genes in C. elegans (Ganko et al. 2003).
To determine if the observed number of LRSs associated with genes is greater or less than what is expected by chance, we computed an expected number of associations based on the probability of an insertion event occurring randomly within a 1- to 1000-bp proximal window or within the transcriptional boundaries of any annotated gene in the genome. The observed number of proximal associations (obs, 82) is not significantly different from what is expected by chance (exp, 85; p > 0.10). In contrast, the observed number of internal associations (obs, 146) is significantly less than what is expected by chance (exp, 382; p < 0.001), presumably due to negative selection.
Consistent with a random distribution model, we found that, as a general rule, those LRS sequences that are most abundant in the genome are also the sequences most frequently associated with genes. There were, however, some notable exceptions. For example, families DM88, GATE, invader 1, and invader 3 have significantly fewer LRS-gene associations than expected (χ2 = 72.3, df = 43, p = 0.003; Table 1) based on the number of family members in the genome. Intrafamily transposon clustering is the likely cause of the low percentage of association in all four of these families. For example, 30 of the 32 DM88 elements are located within a 32-kb stretch of chromosome 3R, and 18 of 26 invader1 elements are located along a separate 28kb stretch of chromosome 3R. Since according to our criteria (see Materials and Methods), only LRSs on the end of an LRS cluster can be scored as being associated with a gene, LRS clustering may explain the reduced associations of DM88, GATE, invader 1, and invader 3 elements.
The Distribution of LRS-Gene Associations Is Not Correlated with Gene Density
While the accumulation of most LRSs does not appear to be tightly correlated with regional gene density (Bartolome et al. 2002; Rizzon et al. 2002), it remains a possibility that those LRSs that are associated with genes may lie within chromosomal regions of high gene density. To test this possibility we determined gene densities across consecutive 200-kb regions of each chromosome. The mean number of genes in each bin was calculated for all regions of the chromosome, then for regions that contained at least one LRS, and, finally, for regions that contained an LRS-gene association. Neither LRS nor LRS-gene associations accumulate in regions significantly more gene dense than the mean gene density of the individual chromosome (Table 2).
To test if LRS-gene associations were more likely to occur between genes with small intergenic distances, we measured the distance from each gene to its neighbor. The overall mean distance from gene to gene (4483 ± 638 bp) is essentially the same as the distance between genes with a proximal LRS (4324 ± 1366 bp, disregarding the LRS). Thus, neither the regional nor the local density of genes is a good predictor of LRS-gene associations.
Most LRSs Located in or Proximal to Genes AreFull-Length or Near-Full-Length in Size
Our results indicate that LRSs associated with genes are significantly larger (5765 ± 178 bp) than the average LRS (4531 ± 242 bp) in the D. melanogaster genome. Since most D. melanogaster full-length retrotransposons are relatively recent insertions (Bowen and McDonald 2001; Kaminker et al. 2002; Lerat et al. 2003) and since Drosophila transposable elements are believed to be actively reduced in size over evolutionary time (Petrov 2002; Petrov and Hartl 1998), our results suggest that most LRS-gene associations are of recent evolutionary origin. This is consistent with recent findings showing that the majority of the full-length or near-full-length LRS-gene associations present in the sequenced Drosophila genome are strain specific (Franchini et al. 2004).
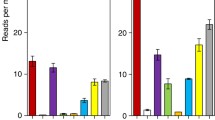
To further investigate whether recent (full-length/near-full-length) insertions are more likely to be associated with a gene than older (small/fragmented) insertions, we looked at the size distribution of all LRSs in the genome. Using a representative consensus sequence from each LRS family as the expected reference size of a full-length element, we found that 348 LRSs are ≥90% of the consensus size (large). Another 123 fragmented LRSs range from 21 to 89% of consensus size (medium), and the remaining 211 LRS are ≤20% of consensus size (small), consisting of 153 small fragments and 58 solo LTRs. When the size distribution of all LRS is compared to the size distribution of LRS associated with genes (Fig. 1), small LRSs were found to be consistently underrepresented (obs, 30; exp, 67), suggesting selection against LRS-gene associations over time. Small LRSs comprise 31.0% of all LRSs in the genome but only account for 17.1% of LRS-gene proximal associations. Small LRSs are even less frequent within genes, accounting for only 11.1% (16/146) of LRSs located within gene boundaries (Fig. 2).

Size distribution of LTR retrotransposons associated with genes. Full-length consensus sizes for individual families were determined from Flybase (Kaminker et al. 2002) or RepBase (Jurka 2000), and the size of each LRS element was compared to the size of the consensus sequence for the appropriate LRS family to calculate a “percentage consensus size.” Full-length/near-full-length LRSs are ≥90% of the consensus size, medium-sized LRS are 21–89% of consensus size, and small LRSs are ≤20% of consensus size. Expected values were calculated based on the distribution of sizes from all 682 LRS in the genome. *Observed values are significantly different from the expected value (p < 0.05).

Percentage of small LTR retrotransposon sequences associated with Drosophila genes. The percentage of small LRSs was computed for all LRSs, for LRSs proximal to a gene (1–1000 bp), for LRSs inside a gene, and for LRSs in or proximal to conserved homologenes.
Large LRSs were found to be associated with genes more frequently (obs, 153; exp, 112) than expected (Fig. 1) based on a random model of association (χ2 = 47.09, p = 0.0006), suggesting either that recent LRS-gene associations are favored by selection and/or, perhaps more likely, that there is a slight preference for LTR retrotransposon insertions in transcriptionally active (open chromatin and/or AT-rich) regions of chromosomes as has been reported in yeast (Sandmeyer et al. 1990).
The LRS size data were further analyzed to determine if the LRS size groups were equally distributed both within genes and proximal to genes. While the relative number of large and medium-sized LRSs varies little within the proximal or internal association groups, the relative number of small LRS proximal to genes is larger than within genes, suggesting that selection is operating against LRSs located within gene boundaries over evolutionary time (Fig. 2).
Functionally Conserved Genes (Homologenes) Are Especially Intolerant of LRS-Gene Associations Over Evolutionary Time
As a general rule, genes involved in basic cellular functions are relatively conserved across taxa, while more recently evolved, specialized genes are taxon specific (e.g., Castillo-Davis et al. 2004; van de Lagemaat et al. 2003). To determine if Drosophila LRSs are differentially associated with these different classes of genes, we analyzed the pattern of LRSs associated with Drosophila genes that have homologues across a broad spectrum of species. Utilizing the 2503 Drosophila genes represented in the NCBI-curated homologene dataset of putative orthologous genes (http://www.ncbi.nlm.nih.gov/HomoloGene/), we identified 51 LRS-homologene associations. The proportion of LRS-homologene associations (51/2503 = 2%) is insignificantly different from the proportion of LRS-gene associations overall (228/13,369 = 1.7%). We found that only 5.9% (3/51) of homologenes were associated with a small, presumably older, LRS (Fig. 2). This value is significantly lower than the frequency of small LRS-gene associations overall (30/228 = 13%). Thus, while newly inserted LRSs (i.e., full-length/near-full-length LRSs) appear to insert in or near homologenes and nonhomologenes with equal frequency, over time, LRS-homologene associations are being preferentially selected against.
Genes Bearing Internal LRSs Are Larger Than Average
Variation in the size of Drosophila genes is primarily due to variation in the size of introns (Holt et al. 2002). Thus, LRSs that insert into large genes might be less likely to disrupt gene function and be eliminated by natural selection. To test if LRSs are preferentially associated with large genes, we compared the mean size of all genes to the mean size of associated genes (excluding the size of the inserted LRS). The results indicate that genes associated with an LRS are three to five times larger than the average Drosophila gene (Fig. 3). Homologenes with an LRS association follow a similar trend. When grouped by associating LRS size, homologenes associated with full-length/near-full-length LRSs are again substantially larger than the average gene (data not shown). Genes associated with small LRSs display tremendous size variation but, on average, are again larger than the typical Drosophila gene (Fig. 3).

Mean size of Drosophila genes with associated LTR retrotransposon sequences. Each bar represents the mean size for a set of genes after disregarding the size of the associated LRS. The two LRS size groups are based on comparisons to consensus size: full-length/near-full-length (LRSs ≥90% of the consensus size) and small (LRSs ≤20% of consensus size). Proximal associations are LRSs within 1–1000 bp of a gene, and internal associations are LRSs inside a gene. Error range indicates 95% confidence interval. *Significantly different gene size for a particular group of genes associated with an LRS compared to the mean size of all genes (p < 0.05).
We found that introns in genes with an internal LRS are more numerous and significantly larger (excluding the size of the insert) than the average-sized intron (Table 3). While exons are more numerous in genes with an internal LRS, they are not significantly larger than the average exon (Table 3). In general, our findings are consistent with the hypothesis that larger genes (with larger/more numerous introns) are more tolerant of LRS insertions. As might be expected, the frequency of LRS insertions proximal to genes is not affected by the size of the associated gene.
Large LRS Are Preferentially Associated with Several Functional Categories of Genes
Several authors have noted that transposons are preferentially associated with certain functional classes of genes (Ganko et al. 2003; Grover et al. 2003; van de Lagemaat et al. 2003). To investigate this question in Drosophila, we grouped our LRS-gene associations using gene ontology (GO) terms. GO terms are descriptors of gene product characteristics hierarchically categorized under three root terms (‘cellular component,’ ‘biological process,’ ‘molecular function’). Using a custom set of Perl scripts, we traced each Drosophila gene descriptor to its respective root term. The cumulative results for all Drosophila genes were used to calculate expectation values for the descriptors of our subset of LRS-associated genes. For large LRS-gene associations, no significant differences were observed between the observed and the expected number of genes encoding cellular component or molecular function (Table 4), but there was a significant deviation from the random expectation (χ2 p = 8.1E-25) for those genes involved in biological processes. Individual analysis of biological process terms (Table 4) demonstrated that the subordinate descriptors ‘development’ (obs, 225; exp, 166; p = 1.4E-07) and ‘behavior’ (obs, 32; exp, 9; p = 1.4E-09) were overrepresented, while the ‘physiological processes’ descriptor was underrepresented (obs, 255; exp, 329; p = 2.5E-09). The subset of homologenes that are associated with LRSs displays a pattern similar to that of associated genes (development, 112 obs/70 exp; behavior, 12/4; physiological processes, 105/149).
We further analyzed the subordinate descriptor terms of the three significant biological processes (Table 5). Significant deviation from expectation was not observed among individual descriptors of the behavior group, though ‘learning and/or memory’ (obs, 10; exp, 5) was twice the expected value. Two development descriptor terms were significantly different. ‘Pattern specification,’ defined as patterns of cell differentiation, was underrepresented (obs, 8; exp, 32; p = 7.0E-08), while ‘morphogenesis’ was overrepresented (obs, 115; exp, 92; p = 1.2E-03). The subordinate descriptor term ‘morphogenesis of an epithelium’ (obs, 13; exp, 4; p = 0.002) was the lone significantly overrepresented morphogenesis term. Two physiological process descriptor terms were also significantly different than expectation. ‘Metabolism’ was underrepresented (obs, 94; exp, 131; p = 1.2E-06), while ‘response to external stimulus’ was overrepresented (obs, 69; exp, 37; p = 1.1E-07). Taken together, large LRSs in Drosophila appear to preferentially associate with genes in select functional groups, including morphogenesis of an epithelium, response to external stimulus, and behavioral functions, while associations with genes involved in metabolism and patterns of cell differentiation are significantly fewer than expected.
Although this observed preference may be due to positive selection, it may also reflect insertional preference. For example, it is known that transcriptionally active genes in an open chromatin configuration may be prone to TE insertions (e.g., Sandmeyer et al. 1990). Since a number of developmental/behavioral genes (e.g., Reinke and White 2002) are known to be transcribed during early stages of development when retrotransposons are transpositionally active (Arkhipova et al. 1995), they may be especially prone to TE insertions. Likewise, since LRS retrotransposons are known to be transcriptionally and transpositionally responsive to external stimuli (Ratner et al. 1992; Strand and McDonald 1985), genes that are also transcriptionally responsive to external stimuli may be especially prone to LRS insertion. Further analyses will be needed to test these hypotheses.
Small LRSs Are Preferentially Associated with Signal Transduction Genes
Only the molecular function group displayed significant differences within the small LRS association dataset (χ2 p = 9.5E-19; Table 4) and a binomial analysis confirmed that ‘signal transduction’ was overrepresented among small LRS associations (obs, 19; exp, 3; p = 1.6E-11). A greater than expected number of signal transduction terms within LRS-associated homologenes was also observed (obs, 14; exp, 4; p = 0.018). This is especially remarkable since signal transduction is underrepresented in the whole homologene set (obs, 194; exp, 307).
Discussion
The recent completion of a number of genome sequencing projects has provided an unprecedented opportunity to investigate the impact of TEs on gene/genome evolution. For example, recent analyses indicated that retrotransposon sequences have contributed to both structural and regulatory gene evolution in humans (e.g., Makalowski 2000; Medstrand et al. 2002; Nigumann et al. 2002). In C. elegans, the available evidence indicates that TEs may have been particularly important in the emergence of recently evolved genes (Ganko et al. 2003). Preliminary comparative analyses of the genomes of chimps and humans suggest that many of the genomic differences between these species are the result of deletions and chromosomal rearrangements mediated by retrotransposons (Britten 2002).
In our study, we have taken a whole-genome approach toward understanding the evolutionary significance of LRSs in Drosophila gene evolution. We found that 248 of the 13,300 genes (1.9%) identified in the sequenced D. melanogaster genome have LRSs proximal to or within transcription boundaries. Of the 682 LRSs present in the D. melanogaster genome, 146 (21.4%) are located within genes, while an additional 82 (12.0%) are located within 1 kb of the 5′ or 3′ gene boundaries. While the number of LRSs located proximal to genes is consistent with what is expected by chance, the number of internal LRS-gene associations is significantly less than expected by chance, indicating that, in general, there is selection against LRS insertions within gene boundaries.
Previous studies have shown that nearly all full-length/near-full-length LRSs in the D. melanogaster genome are recent insertions (Bowen and McDonald 2001; Kaminker et al. 2002; Lerat et al. 2003). This is believed to be due, at least in part, to the fact that processes exist in Drosophila to reduce the size and/or otherwise actively remove TE sequences from the genome (e.g., Moriyama et al. 1998; Petrov 2002; Petrov and Hartl 1998). As a consequence, the relative size of LRSs in Drosophila may be taken as an indicator of the relative time an LRS has been present in the genome. Thus, as a general rule, large, full-length/near-full-length LRSs may be viewed as relatively recent additions to the genome, while smaller fragments may be considered remnants of older insertion events. In light of this distinction, an examination of the numbers and genome distribution of these two size classes of LRS can be useful in gaining insight into the possible action of natural selection on LRS-gene associations over evolutionary time.
Perhaps the most obvious distinction between the distribution of these two size classes of LRS is the relative frequency with which they are associated with genes. The vast majority of LRSs located in proximity to or within genes are full-length/near-full-length elements (Fig. 2). In contrast, there are relatively few small LRSs associated with genes. This result is likely due to the active elimination of LRSs and/or to negative selection against LRS-gene associations over evolutionary time. If the reduction of LRS-gene associations over evolutionary time is independent of selection, no significant difference in the number of LRS-gene associations among different functional classes of genes would be expected. To address this question, we examined the frequency of LRS-gene associations among homologenes relative to all Drosophila genes. Homologenes are genes associated with functions that are generally conserved among even phylogenetically diverse groups of species (Wheeler et al. 2003). Because homologenes encode conserved functions over a broad spectrum of species, they are considered to be older on the evolutionary time scale than genes having homologues in no, or only a few, closely related species. Moreover, because of their broadly conserved functions, homologenes are considered to be relatively less tolerant to genetic change over evolutionary time.
We found that the frequency with which small LRSs are associated with homologenes is significantly lower than the frequency with which they are associated with genes overall. This is consistent with selection operating against LRS sequences associated with homologenes over time and with previous findings in humans (van de Lagemaat et al. 2003) and C. elegans (Ganko et al. 2003) that retrotransposon sequences are preferentially associated with genes encoding more recently evolved functions.
The question remains whether those small LRS-gene associations that have persisted within the Drosophila genome over evolutionary time may be of adaptive significance. To address this question, we examined the functional classification of genes associated with small LRSs and found that the majority of these associations are with nonhomologenes encoding signal transduction functions. Moreover, we found that the frequency of small LRSs associated with genes encoding signal transduction functions is dramatically higher than what is expected by chance. These findings are consistent with the hypothesis that selection has favored the association of small LRSs with Drosophila genes encoding signal transduction functions over evolutionary time. Interestingly, human Alu elements have also been found to be preferentially associated with genes encoding signaling and other rapidly evolving functions in humans (Grover et al. 2003).
There is a growing body of evidence that TE fragments may be a significant contributing factor in the adaptive evolution of Drosophila euchromatic genes encoding signaling and environmentally responsive functions (e.g., Daborn et al. 2002; Franchini et al. 2004; Maside et al. 2002). Additionally, many fragmented LRS-gene associations have recently been identified in D. melanogaster heterochromatin (Dimitri et al. 2003) and evidence exists that at least some LRS-heterochromatic gene associations may be of adaptive significance (McCollum et al. 2002).
Ever since the initial discovery of TEs, there has been considerable debate concerning their adaptive significance. Scientists involved in their discovery generally favored the hypothesis that TEs play an important role in gene regulation and other adaptive functions (e.g., McClintock 1984). Subsequent theoretical demonstrations that TEs may be maintained in populations even in the face of slight negative selection cast considerable doubt on the adaptive hypothesis (e.g., Charlesworth et al. 1997; Doolittle and Sapienza 1980; Hickey 1982; Orgel and Crick 1980). An alternative position is that even if TEs are maintained in populations on a day-to-day basis primarily by nonadaptive processes, they may, over longer spans of evolutionary time, contribute significantly to adaptive gene/genome evolution (e.g., Makalowski 2003; McDonald 1993, 1995). As we have shown that older LRSs are significantly likely to associate with genes of certain functions, the results presented here are consistent with this alternative position.
We have found no unequivocal evidence that recently inserted (full-length/near-full-length) LRSs provide initial adaptive benefit to their host genes. Although this does not preclude the possibility that a particular LRS insertion may be of immediate positive advantage, our results indicate that new LRS-gene associations are, on average, selected against over time. In contrast, when we examined relatively small LRS fragments that have been associated with genes over longer spans of evolutionary time, we found evidence of positive selection, especially with respect to rapidly evolving genes (nonhomologenes) encoding signaling functions. Although it appears that most LRS insertions in or proximal to Drosophila genes are initially either adaptively neutral or of selective disadvantage, over longer spans of evolutionary time, our results are consistent with the hypothesis that small LRS fragments associated with genes have been favored by natural selection.
References
Adams MD, Celniker SE, Holt RA, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287:2185–2195
Arkhipova I, Lyubomirskaya N, Ilyin Y (1995) Drosophila retrotransposons. Landers Press, Austin, TX
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29
Bartolome C, Maside X, Charlesworth B (2002) On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol 19:926–937
Boeke JD, Garfinkel DJ, Styles CA, Fink GR (1985) Ty elements transpose through an RNA intermediate. Cell 40:491–500
Bowen NJ, McDonald JF (2001) Drosophila euchromatic LTR retrotransposons are much younger than the host species in which they reside. Genome Res 11:1527–1140
Britten RJ (2002) Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc Natl Acad Sci USA 99:13633–13635
Brosius J (1999) Genomes were forged by massive bombardments with retroelements and retrosequences. Genetica 107:209–238
Castillo-Davis CI, Kondrashov FA, Hartl DL, Kulathinal RJ (2004) The functional genomic distribution of protein divergence in two animal phyla: coevolution, genomic conflict, and constraint. Genome Res 14:802–811
Celniker SE, Wheeler DA, Kronmiller B, Carlson JW, Halpern A, Patel S, Adams M, Champe M, Dugan SP, Frise E, Hodgson A, George RA, Hoskins RA, Laverty T, Muzny DM, Nelson CR, Pacleb JM, Park S, Pfeiffer BD, Richards S, Sodergren EJ, Svirskas R, Tabor PE, Wan K, Stapleton M, Sutton GG, Venter C, Weinstock G, Scherer SE, Myers EW, Gibbs RA, Rubin GM (2002) Finishing a whole-genome shotgun: release 3 of the Drosophila melanogaster euchromatic genome sequence. Genome Biol 3:RESEARCH0079
Charlesworth B, Langley CH, Sniegowski PD (1997) Transposable element distributions in Drosophila. Genetics 147:1993–1995
Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P, Feyereisen R, Wilson TG, ffrench-Constant RH (2002) A single p450 allele associated with insecticide resistance in Drosophila. Science 297:2253–2256
Dimitri P, Junakovic N, Arca B (2003) Colonization of heterochromatic genes by transposable elements in Drosophila. Mol Biol Evol 20:503–512
Doolittle WF, Sapienza C (1980) Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603
Franchini LF, Ganko EW, McDonald JF (2004) Retrotransposon-gene associations are wide-spread among D. melanogaster populations. Mol Biol Evol:msh116
Ganko EW, Bhattacharjee V, Schliekelman P, McDonald JF (2003) Evidence for the Contribution of LTR Retrotransposons to C. elegans Gene Evolution. Mol Biol Evol 20:1925–1931
Grover D, Majumder PP, Rao CB, Brahmachari SK, Mukerji M (2003) Nonrandom distribution of alu elements in genes of various functional categories: insight from analysis of human chromosomes 21 and 22. Mol Biol Evol 20:1420–1424
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32(Database Issue):D258–D261
Hickey DA (1982) Selfish DNA: a sexually-transmitted nuclear parasite. Genetics 101:519–531
Holt RA, Subramanian GM, Halpern A, et al. (2002) The genome sequence of the malaria mosquito Anopheles gambiae. Science 298:129–149
Hoskins RA, Smith CD, Carlson JW, Carvalho AB, Halpern A, Kaminker JS, Kennedy C, Mungall CJ, Sullivan BA, Sutton GG, Yasuhara JC, Wakimoto BT, Myers EW, Celniker SE, Rubin GM, Karpen GH (2002) Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol 3:RESEARCH0085-5
Jordan IK, Rogozin IB, Glazko GV, Koonin EV (2003) Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet 19:68–72
Jurka J (2000) Repbase update: a database and an electronic journal of repetitive elements. Trends Genet 16:418–420
Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, Ashburner M, Celniker SE (2002) The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol 3:RESEARCH0084-4
Leeton PR, Smyth DR (1993) An abundant LINE-like element amplified in the genome of Lilium speciosum. Mol Gen Genet 237:97–104
Lerat E, Rizzon C, Biemont C (2003) Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res 13:1889–1896
Li WH, Gu Z, Wang H, Nekrutenko A (2001) Evolutionary analyses of the human genome. Nature 409:847–849
Makalowski W (2000) Genomic scrap yard: how genomes utilize all that junk. Gene 259:61–67
Makalowski W (2003) Genomics. Not junk after all. Science 300:1246–1247
Maside X, Bartolome C, Charlesworth B (2002) S-element insertions are associated with the evolution of the Hsp70 genes in Drosophila melanogaster. Curr Biol 12:1686–1691
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
McCollum AM, Ganko EW, Barrass PA, Rodriguez JM, McDonald JF (2002) Evidence for the adaptive significance of an LTR retrotransposon sequence in a Drosophila heterochromatic gene. BMC Evol Biol 2:5
McDonald JF (1993) Evolution and consequences of transposable elements. Curr Opin Genet Dev 3:855–864
McDonald JF (1995) Transposable elements: possible catalysts of organismic evolution. Trends Ecol Evol 10:123–126
Medstrand P, Landry JR, Mager DL (2001) Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J Biol Chem 276:1896–1903
Medstrand P, van de Lagemaat LN, Mager DL (2002) Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res 12:1483–1495
Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, Smith CD, Tupy JL, Whitfied EJ, Bayraktaroglu L, Berman BP, Bettencourt BR, Celniker SE, de Grey AD, Drysdale RA, Harris NL, Richter J, Russo S, Schroeder AJ, Shu SQ, Stapleton M, Yamada C, Ashburner M, Gelbart WM, Rubin GM, Lewis SE (2002) Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol 3:RESEARCH0083
Moriyama EN, Petrov DA, Hartl DL (1998) Genome size and intron size in Drosophila. Mol Biol Evol 15:770–773
Nekrutenko A, Li WH (2001) Transposable elements are found in a large number of human protein-coding genes. Trends Genet 17:619–621
Nigumann P, Redik K, Matlik K, Speek M (2002) Many human genes are transcribed from the antisense promoter of l1 retrotransposon. Genomics 79:628–634
Orgel LE, Crick FHC (1980) Selfish DNA: the ultimate parasite. Nature 284:604–607
Papatsenko DA, Makeev VJ, Lifanov AP, Regnier M, Nazina AG, Desplan C (2002) Extraction of functional binding sites from unique regulatory regions: the Drosophila early developmental enhancers. Genome Res 12:470–481
Petrov DA (2002) DNA loss and evolution of genome size in Drosophila. Genetica 115:81–91
Petrov DA, Hartl DL (1998) High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol 15:293–302
Ratner VA, Zabanov SA, Kolesnikova OV, Vasilyeva LA (1992) Induction of the mobile genetic element Dm-412 transpositions in the Drosophila genome by heat shock treatment. Proc Natl Acad Sci USA 89:5650–5654
Reinke V, White KP (2002) Developmental genomic approaches in model organisms. Annu Rev Genomics Hum Genet 3:153–178
Rizzon C, Marais G, Gouy M, Biemont C (2002) Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res 12:400–407
Sandmeyer SB, Hansen LJ, Chalker DL (1990) Integration specificity of retrotransposons and retroviruses. Annu Rev Genet 24:491–518
Strand D, McDonald JF (1985) Copia is transcriptionally responsive to environmental stress. Nucleic Acids Res 13:4401–4410
van de Lagemaat LN, Landry JR, Mager DL, Medstrand P (2003) Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet 19:530–536
Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L (2003) Database resources of the National Center for Biotechnology. Nucleic Acids Res 31:28–33
Acknowledgments
This research was supported by a National Institutes of Health (NIH) Grant to J.F.M. E.W.G. was supported through an NIH Genetics Training Grant. We thank Paul Schliekelman (University of Georgia) for statistical advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Juergen Brosius
Rights and permissions
About this article
Cite this article
Ganko, E.W., Greene, C.S., Lewis, J.A. et al. LTR Retrotransposon-Gene Associations in Drosophila melanogaster. J Mol Evol 62, 111–120 (2006). https://doi.org/10.1007/s00239-004-0312-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0312-4