
Abstract
Genomes of prokaryotes harbor genomic islands (GIs), which are frequently acquired via horizontal gene transfer (HGT). Here I present an analysis of GIs with respect to gene-encoded functions. GIs were identified by statistical analysis of codon usage and clustering. Genes classified as putatively alien (pA) or putatively native (pN) were categorized according to the COG database. Among pA and pN genes, the distribution of COG functions and classes were studied for different groupings of prokaryotes. Groups were formed according to taxonomical relation or habitats. In all groups, genes related to class L (replication, recombination, and repair) were statistically significantly overrepresented in GIs. GIs of bacteria and archaea showed a distinct pattern of preferences. In archeal GIs, genes belonging to COG class M (cell wall/membrane/envelope biogenesis) or Q (secondary metabolites biosynthesis, transport, and catabolism) were more frequent. In bacterial GIs, genes of classes U (intracellular trafficking, secretion, and vesicular transport), N (cell motility), and V (defense mechanisms) were predominant. Underrepresentation was strongest for genes belonging to class J (translation, ribosomal structure, and biogenesis). Among single COG functions overrepresented in GIs were transferases and transporters. In both superkingdoms, HGT enhances genomic content by meeting demands that are independent of the studied habitats. These findings are in agreement with the complexity theory, which predicts the preferential import of operational genes. However, only specific subsets of operational genes were enriched in GIs. Modification of the cell envelope, cell motility, secretion, and protection of cellular DNA are major issues in HGT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microbial genomes contain genomic islands (GIs), i.e., stretches of DNA exhibiting a conspicuous composition with regard to sequence or encoded functions. Frequently in these clusters, the number of hypothetical proteins is high or encoded functions are highly specific, often enhancing the fitness of the species. Typical examples are genes mediating antibiotic resistance, genes encoding transport systems, and gene clusters related to pathogenicity (Hacker and Kaper 2000). Possible sources for GIs are bacteriophages: the genome of Escherichia coli K-12 contains 11 (Ohnishi et al. 2001); the genome of Bacillus subtilis, 10 putative prophages (Kunst et al. 1997). There is evidence that GIs were predominately acquired via horizontal gene transfer (HGT) (Ochman et al. 2000). This process is regarded as an important force for the enhancement of genetic diversity. HGT adds new genes to a genome, often originating from taxonomically unrelated species. A recent study (Nakamura et al. 2004) showed that gene-encoded functions related to cell surface, DNA binding, and pathogenicity were overrepresented in GIs. These functions are frequently fulfilled by housekeeping, i.e., operational genes. An explanation why operational genes undergo HGT more frequently than informational ones is proposed by the complexity hypothesis (Jain et al. 1999): Informational genes are typically members of large and complex systems making the exchange among genomes rather unlikely.
In the following, I introduce an analysis of GI composition determined for 63 prokaryotic genomes. It addresses the following two questions: Which gene-encoded functions are enriched in bacterial and archeal GIs, respectively? and Does lifestyle or habitat influence the composition of GIs? In addition, the analyses are used to test the complexity hypothesis introduced to explain GI composition: If the theory holds, the composition of GIs is similar with respect to the characteristics of protein interactions, irrespective of additional factors. It is shown that the gene content of bacterial and archeal GIs is significantly different. However, colonization of ecological niches does not markedly influence GI composition. Genes enriched in GIs are a specific subset of housekeeping genes; dominating protein functions are frequently related to cell envelope biosynthesis, cell motility, secretion, or DNA protection.
Material and Methods
Material
The content of the COG database (Tatusov et al. 1997, 2003) was utilized. The current version classifies the genes of 63 prokaryotic species into 4873 groups named COGs. Each COG is a set of genes from different species that code for the same function. COGs are organized in 25 functional categories defining the role of encoded proteins (see Table 1). Genomes were grouped according to lifestyle or taxonomical relation (see Table 2).
Analyzing GI Composition
Subsets of the considered genomes were used to compile species-groups. Then GIs were predicted using the program SIGI (Merkl 2004). For each gene, the COG classification (the term class is used synonymously in the following) was determined and according to SIGI’s prediction, added to histograms summing the COG classes for putatively alien (pA) and putatively native (pN) genes. If the number of COGed pA genes was >200, the species-group was further processed. Two histograms were generated, storing the absolute frequencies #pA k and #pNk for each COG class k. Absolute frequencies were the basis for calculating relative frequency values f(pA k ), f(pN k ) and ratios r(k) = f(pA k ) / f k (pN k ). For each COG class k, the absolute number of expected pA gene exp #pA k was determined as
A chi-square test was computed for the set of pairs {#pA k , exp#pA k | k = 1,...,25}. Species-groups were regarded as having a statistically significantly skewed GI composition, if the test was significant at the 1% level.
Evaluating the Occurrence of Individual Gene-Encoded Functions or COG Classes
For a COG function CF, its occurrence in GIs (#pA CF ) and in the rest of the genomes (#pN CF ) was determined. A chi-square test was computed considering the four values #pA CF , #pN CF , and the totals of pA or pN genes determined for a species-group. A COG function was considered statistically over- or underrepresented, if the chi-square test was significant at the 1% level. The skew of an individual COG class k was tested analogously by using values #pA k and #pN k .
Results
In order to study protein functions accumulated in GIs, two problems have to be solved. In groups of genomes, compiled according to phylogenetic relationship or with respect to habitats, (i) GIs have to be identified and (ii) gene-encoded functions have to be categorized. A categorization facilitates statistical analyses and gives a more general view of GI composition.
Categorizing Gene-Encoded Functions in GIs
The program SIGI (Merkl 2004) was used to identify GIs. It is based on the genome theory (Grantham et al. 1980) and the Neyman–Pearson lemma and computes log-odds ratios in order to analyze codon usage of each gene in multiple pairwise comparisons. Codon usage of each gene was compared to 371 samples deduced from the CUTG database (Nakamura et al. 1999), which represents microbial codon usage. For statistical reasons, SIGI did not consider genes shorter than 100 codons. In an iterative approach, a cutoff was chosen dynamically which decided on the classification of a gene as being putatively native (pN) or putatively alien (pA). A gene was predicted as alien, if one of the log-odds ratios exceeded the cutoff. SIGI predicted only clusters containing at least five consecutive pA genes as GIs; shorter clusters were dropped. During SIGI’s design phase, it has been demonstrated that this heuristic is at least as sensitive as other methods exploiting compositional complexity. SIGI had identified compositional atypical gene clusters with similar efficiency as hidden Markov models did (see Merkl 2004).
To classify and to categorize gene functions, information stored in the COG database was utilized. The database is built on the concept of orthology and consists of genes grouped into COGs. Each entry of the database (a cluster of orthologous groups of genes, a COG) is a set of genes from different species that code for the same function. COGs are organized in 25 functional categories (also named classes in the following) defining the role of encoded proteins. Examples of such categories are “translation, ribosomal structure, and biogenesis” and “cell motility.” Table 1 is a listing of all categories. To date, the genes of 63 prokaryotic species are classified in 4873 COGs. A prerequisite for adding a gene to a COG is that the encoded protein sequence shows significant similarity to gene products of two other species. Thus, a minimal COG contains three genes from phylogenetically distinct species. Due to this concept, the fraction of genes that belong to COGs varies among genomes. On average, 70% to 75% of the protein-coding genes of a genome are annotated in the COG database. All 63 prokaryotic genomes included in the COG database were analyzed using SIGI. For each gene, the COG classification was determined and according to SIGI’s prediction, added to histograms summing the COG classifications for pA and pN genes. For each of the considered species-groups, the number of COGed pA genes was counted. For statistical reasons, only groups having more than 200 such genes were further processed (all listed in Table 2). For each of these species-groups, two histograms were generated, storing the absolute frequencies #pA k and #pN k for all COG classes k. Absolute frequencies were used to calculate relative frequency values f(pA k ), f(pN k ), and ratios r(k) = f(pA k ) / f(pN k ).
An example illustrates best the determination of relative frequencies and ratio values. If we consider for a COG class L #pA L = 272 and #pN L = 1324, then we get f(pA L ) = 272/972 = 0.280, f(pN L ) = 1324 /22,914 = 0.058, and r(L) = f(pA L )/f(pN L ) = 0.280/0.058 = 4.84 (see numbers for COG class L of data set “archaea” in Table 3). Here, 972 is the sum of all pA genes and 22,914 is the sum of all pN genes determined in the data set “archaea.” This ratio value indicates that genes assigned to COG class L are 4.8 times as frequent in archeal GIs as among pN genes.
The conversion to relative frequencies is a necessary step if one wants to compare relative abundances of COG classes among pA and pN genes or between different species-groups. As additional parameters and for both classes (pA, pN) the fraction of genes which were not elements of the COG database and the fraction of hypothetical genes were determined and added to Tables 3 and 4, together with the respective ratio values. A chi-square test (significance level set to 1% for all analyses described below, if not otherwise noted) was computed on absolute gene numbers (#pA, expected #pA) in order to identify relevant results. Statistically significant differences in the distribution of COG classes were identified in archaea, Euryarchaeota, Crenarchaeota, hyperthermophilic archaea, obligately anaerobic archaea, bacteria, Gram-plus (low-GC, Gram-positive bacteria), Proteobacteria, the gamma subdivision, mesophilic, acidophilic, and aerobic bacteria, facultatively aerobic/anaerobic bacteria, and opportunistic pathogens and symbionts. Representative results are compiled in Tables 3 and 4 and plotted in Figs. 1 and 2. Genes sorted into COG classes Y and Z did not occur (see Tables 3 and 4); respective ratios are not plotted. Ratio values r(k) (see Tables 3 and 4) indicated that few categories were overrepresented in GIs: Genes related to class L (replication, recombination, and repair) were abundant in the GIs of all groups. This effect is due to SIGI’s classification of a great number of genes related to DNA translocation like transposases and integrases as pAs. In archaea, genes belonging to class M (cell wall/membrane/envelope biogenesis) or to class Q (secondary metabolite biosynthesis, transport, catabolism) were fourfold and twofold, respectively, as frequent as they appeared among pN genes. The overrepresentation of class L in Crenarchaeota was even more distinct than in Euryarchaeota (see Table 3). The underrepresentation of gene-encoded functions was in archeal GIs strongest for the classes H (coenzyme transport and metabolism), J (translation, ribosomal structure, and biogenesis), and F (nucleotide transport and metabolism). For 10 COG classes, the ratio value was ≤0.5, indicating an at least twofold underrepresentation of related gene-encoded functions in archeal GIs. In general, the skew was stronger than in bacterial GIs (see below). The composition of GIs determined for hyperthermophilic or obligately anaerobic archaea did not differ noticeably from the average archeal GI (see Table 3 and Fig. 2). Although these species-groups overlap, the histograms indicate that the preferences among genes acquired by HGT are similar, even in extreme environments.

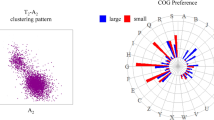
Composition of archeal and bacterial GIs with respect to COG classes. A For archeal and bacterial genomes as listed in Table 2, ratio values r arch (k) or r bact (k) as given in Tables 3 and 4 were plotted for each COG class k. For abbreviations of COG classes, see Table 1. B The weighted logarithm of respective quotients r arch (k)/r bact (k) was plotted for each COG class k. The factor m was set to #pA Bact /300 in order to scale the quotient with the absolute number of bacterial pA genes. The quotient for class W was set to 0 because of the small sample size; compare Tables 3 and 4.

Composition of GIs for different species-groups of microbes. Microbial genomes were grouped according to Table 2, and ratio values r(k) as given in Table 3 or Table 4 were plotted. For each COG classification k the quotient of ratio values r(k) = f(pA k )/f(pN k ) (compare Tables 3 and 4) was calculated. Groups were selected according to phylogenetic relation or lifestyle. A ratio value r(k) of 4 means that gene-encoded functions subsumed in COG class k are in GIs four times as frequent as among putative native genes.
In bacterial GIs (see Table 4 and Fig. 1A), and in addition to the overrepresentation of class L, genes related to class U (intracellular trafficking, secretion, and vesicular transport), N (cell motility), W (extracellular structures), or V (defense mechanisms) were more frequent. This bias was even more pronounced in GIs of opportunistic pathogens and symbionts (and parallel in the gamma subdivision of Proteobacteria, which includes most of the species constituting the species-set PS). The underrepresentation in bacterial GIs was strongest for the COG classes J (translation, ribosomal structure, and biogenesis), F (nucleotide transport and metabolism), and D (cell cycle control, cell division, chromosome partitioning). Interestingly, the skew was less pronounced than in archeal GIs, r(J) and r(F) were the only two ratio values <0.5.
Again, lifestyle did not noticeably influence the distribution of COG classes. The composition of GIs for aerobic bacteria and for facultatively aerobic/anaerobic bacteria was similar to the mean bacterial case. The differences seen for class W (extracellular structures) were presumably due to the small sample sizes. This observation indicates the limitations of this approach. An even finer granularity of gene-encoded function or taxonomical decomposition is not feasible for statistical reasons.
For a direct comparison of the skews seen in archaea and bacteria, a ratio comp was computed as the logarithm of respective quotients r arch (k)/ r bact (k) multiplied with the weight m and plotted for each COG class k (Fig. 1B). The factor m was set to #pA Bact /300 in order to relate the quotient to the absolute number of bacterial pA genes. For the comparison, the absolute size of the comp value is less important; of interest is its sign. If the ratio value r arch (k) is larger than r bact (k), which indicates a stronger overrepresentation of COG class k in archeal GIs, the log ratio is positive; otherwise it is negative. The above findings clearly demonstrate that archaea and bacteria acquired different protein functions via HGT.
In order to assess the complexity hypothesis, the occurrence of informational and operational genes in GIs was studied. Definitely, COG classes J (translation, ribosomal structure, and biogenesis) and K (transcription) subsume informational genes, whereas E (amino acid transport and metabolism) and M (cell wall/membrane/envelope biogenesis) contain operational genes. The ratio values of class J were ≤0.45 in all groups, but ratio values of class K varied in the range between 0.59 and 1.46. However, single COG functions related to class K and overrepresented in GIs could not be classified as informational genes in the strict sense. These were mostly transcriptional regulators (see below). Ratios of class E were ≤0.69, and those of class M ≥1.27. In summary, these results are consistent with the notion that specific subgroups of operational genes were preferentially accumulated in GIs.
COG Functions Representative of GIs
The categorization of gene-encoded functions allows a general characterization of GI composition. For a more specific analysis of GI content, the distribution skew of individual COGs was determined. For each COG, the number of genes identified as pA or pN was counted in order to identify those COG functions overrepresented in GIs of each of the groups introduced in Table 2 and compared to the respective sum of all pA or pN genes. Statistical significance was assigned with a chi-square test. Only those COGs were further processed that showed a significant imbalance (p < 0.01 for each set) in their distribution among pA and pN genes. Table 5 gives a representative example. It lists all COGs which occurred at least five times in archeal GIs and showed such a skewed distribution. Nearly half of the COG functions were related to class L (DNA replication, recombination and repair). Nearly a quarter of the genes was classified as belonging to class R (general function prediction only) or S (function unknown). Three hits were glycosyltransferases (COG0436, COG0438, and COG1216), one was a SAM-dependent methyltransferase (COG0500), and one an acetyltransferase (COG0454). The latter enzymes were overrepresented in the GIs of nearly all studied groups. In addition, DNA restriction/modification systems were frequently found in GIs. These combinations consist of a methyltransferase (like COG0500, SAM-dependent methyltransferases) and a restriction enzyme (like COG0732 or COG1403). These systems act as a defense against infection by bacteriophages or foreign DNA and protect DNA against attack via restriction endonucleases (for a review see Jeltsch 2002).
Distribution asymmetry of several gene-encoded functions was quite drastically. Individual skews have to be compared to the ratio value deduced from all pA and pN genes in archaea, which is 972/22,914 = 0.04 (see Table 3). The distribution skew, e.g., of COG0438 (a glycosyltransferase) is 0.53 and results from the quotient 40/76 = 0.53, which is the ratio of COG0438 genes being classified as pA or pN. These two ratio values differ by a factor of 12, which indicates a 12-fold enrichment of this gene-encoded function in archeal GIs. Even the last entry in Table 5, COG1277 (element of an ABC transporter), is more than sixfold enriched in archeal GIs. In the following, selected COG functions overrepresented in bacterial GIs and not classified as belonging to class L, R, or S are given together with their rank (in parentheses), resulting from a sorting on chi-square values determined for statistical analysis (p ≤ 0.01 in all cases). COG functions overrepresented in bacterial GIs were: COG3209 (4) Rhs family protein, COG3539 (8), COG3121 (14), and COG3188 (19) P pilus assembly proteins, COG4669 (31), COG4790 (34), COG4791 (37), COG4789 (42), and COG4794 (50) components of Esc, a type III secretory pathway, COG1450 (55) component PulD of a type II secretory pathway, COG1396 (10) and COG3311 (38) predicted transcriptional regulators, COG0732 (27) restriction endonuclease S subunits, COG0438 (28) glycosyltransferases, COG1442 (29) lipopolysaccharide biosynthesis proteins, LPS:glycosyltransferases, COG0662 (39) mannose-6-phosphate isomerase, COG2771 (44) DNA-binding HTH domain-containing proteins, COG3617 (68) prophage antirepressor, and COG1403 (69) restriction endonuclease. These examples show that among those proteins classified in the COG database, secretory systems and enzymes involved in cell wall biosynthesis or DNA modification are most frequent in bacterial GIs.
COG class K “transcription” was assumed to contain informational genes (see above). According to the complexity theory, informational genes are no candidates for HGT. However, in bacterial GIs, an overrepresentation of class K genes was observed (see above and Table 4). In all bacterial species-groups the ratio value was >1.0. The highest contribution to the overrepresentation of class K in bacterial GIs was assigned to the following genes (the list gives gene-encoded functions and, in parentheses, ranks which were determined as above): COG1396 (10), COG3311 (38) and COG0583 (85) transcriptional regulators, COG2771 (44) DNA-binding HTH domain-containing proteins, COG2207 (49) AraC-type DNA-binding domain-containing proteins, COG3710 (53) DNA-binding winged-HTH domains, and COG3617 (68) prophage antirepressor. These gene-encoded functions are not involved in those complex interactions that are characteristic for informational genes. In contrast, sigma factors which are assumed to interact in a complex manner with RNA polymerases are rather underrepresented in GIs (see below).
Genes occurring in only one of several, phylogenetically closely related species are candidates for horizontal gene transfer. For an application of this approach used to identify horizontally transferred genes, see for example Makarova and Koonin (2003). An analysis of GIs predicted by SIGI with respect to phyletic patterns revealed no clear picture. GIs were often an intermixture of three kinds of genes, namely, genes not represented in the COG database, genes specific for a small group of taxonomically related species, or orthologous genes found in a broad range of species. Tables 6 gives a typical example showing the composition of a GI identified in the genome of the archaea Halobacterium. Each line characterizes a pA gene. The plotted phyletic pattern lists the occurrence of the gene function in those microbial genomes that contribute to the COG database. A − indicates that the gene function does not occur in the respective genome. Glycosyltransferases are involved in the biosynthesis of the lipopolysaccharide which is an important element of the outer membrane of Gram-negative bacteria. This fact explains why the last four positions of the phyletic pattern are blank, which indicates the occurrence of gene-encoded functions in the group GP (low-GC, Gram-positive bacteria). The family of glycosyltransferases (COG0438) represented three times in the depicted sample was abundant in GIs and found in nearly all species contributing to the COG database. COG0438 is an example of a gene whose “alienism” could not be derived from an analysis of the phyletic pattern implicitly deposited in the COG. The findings presented here argue for a massive exchange of these enzymes via HGT.
Transcriptional Regulators Which AreUnderrepresented in GIs
The complexity hypothesis predicts that transcriptional regulators that interact in a complex manner with RNA polymerases are less likely subject to HGT (see Borukhov and Nudler [2003] for a recent review on the holoenzyme). Among bacterial genomes, SIGI predicted for sigma factors the following ratios of pA/pN genes: COG0568 sigma 70, sigma 32 1/96, COG1191 sigma subunit 1 1/44, COG1508 sigma54 homologue 1/29, COG1595 sigma24 homologue 8/146, and COG4941 predicted sigma factor 0/8. For all sigma factors, the mean ratio is 11/323 = 0.03, which is half of the mean pA/pN determined for bacterial genes. A chi-square test gives p = 0.07 for all sigma factors and p = 0.05 for sigma 70, which is the major factor in transcriptional activation. This result shows that sigma 70 is significantly underrepresented in GIs. For COG1396, a putative transcriptional regulator which is significantly overrepresented in GIs, this ratio is 82/251 = 0.32. The two ratio values differ by a factor of 10, which indicates the broad range of distribution skews found even among proteins with similar functions.
Discussion
Each analysis of HGT raises the question whether its approach of identifying horizontally transferred genes is valid. Therefore, arguments concerning the methodology used here will be discussed first.
Evaluating SIGI’s Potential for GI Prediction
A variety of methods has been developed to identify GIs. The underlying concepts are based on the analysis of sequence composition (Abe et al. 2003; Karlin 2001; Lawrence and Ochman 1998; Tu and Ding 2003; Wang et al. 2001) or gene neighborhood (Ruepp et al. 2000), on phylogenetic studies (Nesbø et al. 2001), or on a combination of these approaches (Garcia-Vallvé et al. 2000; Nesbø and Doolittle 2003). For a comparison, see Ragan (2001a, b) also for studies not considered here. Tools have been generated that tabulate GIs (Garcia-Vallvé et al. 2003; Mantri and Williams 2004) or that allow the comparison of genomes (Florea et al. 2003). However, all these methods have their specific drawbacks, and due to the differing results achieved for the same genomes, the quantification of HGT is still a matter of debate (Doolittle 1999; Kurland et al. 2003). It might be that each method identifies a different class of alien genes (Lawrence and Ochman 2002; Ragan 2001b).
It has been argued that approaches based on codon usage analysis generate large numbers of false-positive or false-negative hits. A strange and atypical DNA composition may be due to location, strand, or genomic content. Mutational effects could be replication or transcription dependent. It is impossible, to exclude the impact of such mutational forces on the prediction of GIs. However, it has been shown that compositional analyses leave out only few transfer events (Daubin et al. 2003). This finding weakens the above objection referring false-negative hits. The risk of generating false-positive hits can be reduced by focusing on the prediction of pA gene clusters. Such an approach elegantly exploits and combines biological evidence and statistical principles: As has been established, genomic islands frequently have a size of 10–200 kb (Hacker and Kaper 2000). This is an important premise, because the probability of predicting false positives decreases drastically for gene clusters consisting of n genes from p(A) to p(A)n, if p(A) is the probability of predicting a horizontally transferred gene. Even for a high probability value of p(A) = 0.3 and a small number of n = 5 genes, p(A)n is <0.0025. A rough estimation (1/ p(A)n) gives that then approximately 1 of 400 clusters occurs merely by chance and is a false classification. This crude estimate, which judges predictions as being independent of the context, is presumably imprecise but illustrates a correct trend. Therefore, cluster analysis has been considered in the design of recently introduced algorithms (Merkl 2004; Nakamura et al. 2004).
There is a second line of arguments that lends additional support to algorithms analysing compositional complexity. The assumption that the latter methods might overlook genes acquired by horizontal transfer could be valid for more ancient events, which were subject to an amelioration process (Lawrence and Ochman 1997) for a longer time period. Recently acquired genes were detected to a great extend by methods based on compositional complexity (Daubin et al. 2003; Ragan 2001b). Lawrence and Ochman (1998) estimated the age of imported genes and concluded that most were relatively recent, i.e., acquired within the last few million year (see, e.g., de la Cruz and Davies 2000). This suggests that older imports have been purged from the genomes, presumably because these genes did not improve fitness (Kurland et al. 2003). Given this reasoning, there is no need to search for huge amounts of ancient pA genes.
Concerning SIGI, it has been shown that its predictions overlapped to a great extent with those generated by other methods and that previously assigned GIs were correctly identified (Merkl 2004). SIGI assesses for its prediction variations in the usage of each individual codon among microbial species and does not interpret the amino acid sequence level. Therefore, it is highly unlikely that protein function, which might modulate the amino acid composition as, e.g., in membrane-bound proteins, might influence SIGI’s classifications. It is valid to expect a classification into pA and pN genes independent of protein function or COG classification. Interestingly, a recent analysis of genomewide codon bias gave further support to Grantham’s hypothesis of a specific codon usage in individual genomes and suggests that mutational processes are the main factors shaping codon bias (Chen et al. 2004). This finding lends additional support to algorithms like SIGI, which predict GIs based on codon usage analysis.
Biological Functions Dominating GIs
GIs harbored few informational genes. Genes related to class K and accumulated in GIs were not informational genes stricto sensu. Among operational genes, specific subgroups were preferentially accumulated in GIs: In addition to enzymes needed for DNA translocation, only a few functional classes were overrepresented. These were frequently enzymes involved in the biogenesis of the outer membrane. A prominent example was COG function 438. These glycosyltransferases were present in all species besides those constituting group GP (low-GC Gram-positive bacteria). Glycosyltransferases are frequently involved in the biosynthesis of the lipopolysaccharide (LPS) located in the outer membrane. LPS is an important element of the outer membrane of Gram-negative bacteria. Modifications of LPS composition may have drastic consequences for the cell with respect to resistance against bacteriophages and antimicrobial compounds. The acquisition of a new gene cluster for LPS biosynthesis in Vibrio cholerae has been considered a major reason for the cholera epidemic occurring 1992 in India (Mooi and Bik 1997). LPS is an important virulence factor (Dow et al. 1995) and induces resistance in plants against pathogens. Extreme variation in LPS gene cluster composition has been reported in animal pathogenic bacteria. Eleven highly divergent gene clusters have been shown to constitute a LPS-specific locus in Pseudomonas aeruginosa, an opportunistic human pathogen (Raymond et al. 2002). COG0438 describes an enzyme function encoded several times, presumably with different specificity in many genomes. The overrepresentation illustrated above is in agreement with the occurrence of putative glycosyltransferases in GIs of P. aeruginosa (Karlin 2001) and Deinococcus radiodurans (Karlin and Mrázek 2001). The E. coli K-12 protein RfaG (gene rfaG is a member of COG0438) is involved in LPS biosynthesis; rfaG mutants exhibit a lower level of expression of some outer membrane proteins (Genevaux et al. 1999; Raetz and Whitfield 2002). A comparison of the rfa/waa region, occurring in E. coli K-12, E. coli EDL933, and Shigella flexneri 2a, showed that this region was present in almost-identical form in the genomes of S. flexneri and of E. coli EPL933, whereas the majority of the E. coli K-12 genes exhibited less similarity to the former ones (Jin et al. 2002). These findings exemplify the character and the dynamics of gene content occurring in GIs. Modification of the outer membrane is a major selective factor for acquiring DNA. Results compiled in Table 5 confirm this conclusion also for archaea. A second incentive for the acquisition of novel genes is the defense against infection by bacteriophages or mobile transposons and the protection of DNA against attack via restriction endonucleases, as provided by methyltransferases and restriction enzymes.
An alternative explanation of the results presented above is the assumption that some genes are less subject to HGT because the recipient’s genome contains already orthologs. Orthologous replacements are assumed to be rare and orthologous pA genes are presumably hard to detect. Most genes involved in translation or replication are universal and would thus be subject to orthologous transfer, which might explain their underrepresentation in GIs. However, the following has to be considered: The complexity hypothesis (Berg and Kurland 2002; Jain et al. 1999; Kurland 2000) predicts, for transcriptional regulators involved in complex interactions with RNA polymerases, a lower frequency of HGT. The above findings concerning sigma factors are in agreement with this prediction.
Recently a study was presented which focuses on a global view on biological function of horizontally transferred genes in prokaryotic genomes (Nakamura et al. 2004). For the categorization of gene-encoded functions, the authors used the TIGRFAM database (Haft et al. 2003) and studied for pA genes their role and subrole categories which constitute the categorization scheme for TIGRFAM. For the identification of pA genes the authors introduced a method which analyzes the compositional complexity of DNA fragments. For a sample of 166 prokaryotic genomes, 14% of the genes were on average predicted as being horizontally transferred. For 11 species, at least 20% of the genes were identified as pA. Nicolas et al. 2002) used a hidden Markov model (HMM) for the identification of compositional atypical segments of the B. subtilis genome. From the data deposited at http://www.poplar.genes.nig.ac.jp/∼hgt/, it can be deduced that approximately half of the pA genes predicted by Nakamura et al. for the B. subtilis genome do not belong to a potential horizontal transfer region identified by the HMM. This observation indicates that the findings deduced by Nakamura et al. may at least partially depend on the prediction of pA genes lying isolated or in short pA clusters. SIGI uses—as explained above—a more conservative approach in predicting GIs only. In addition, the concept presented here relates the occurrence of gene-encoded functions in GIs to their frequency in the rest of the genome. Despite these differences in methodology, findings which concern global trends presented by Nakamura et al. (2004) and above are consistent. In both studies, an overrepresentation of gene-encoded functions related to plasmids, phages, transposon functions, and cell envelope was identified. The algorithm of Nakamura et al. identified as pA many gene-encoded functions from the TIGR role “regulatory functions” and identified for pAs an overrepresentation of the subrole “DNA interactions.” As shown above, the occurrence of transcriptional regulators in GIs is biased. In bacterial GIs, some regulators are significantly overrepresented, like COG functions 1396 and 3311; however, sigma factors are underrepresented. Due to the different intentions of the studies (global trends vs. specific demands), the particular approaches used for the identification of pA genes, and the two different classification schemes, it is difficult to discuss the results in more detail.
Phyletic Differences in GI Composition
Compared to bacteria, archeal GIs harbored more functions related to cell envelope biogenesis (Fig. 1B). This overrepresentation of related gene-encoded functions was seen even in GIs of hyperthermophilic archaea, populating an ecological niche assumed to be less competitive. The composition of the cell wall is a feature distinguishing archaea from bacteria. A monolayer of glycosylated S-layer proteins serves as an envelope for the archeal cell. The glycosylation machinery of archaea is complex; it is assumed that glycosylation occurs at the outer cell surface. The machinery is therefore similar to the eukaryotic one (see, e.g., the review by Eichler 2003). However, the data presented above argue for a massive horizontal transfer of glycosyltransferases involved in cell envelope biogenesis. The phyletic pattern of COG0438 presented in Table 6 suggests for archaea the exchange of enzymes for cell wall modification at least to some extent with bacteria.
In contrast to archeal GIs, bacterial ones did harbor more gene-encoded functions related to cell motility, transcription, intracellular trafficking, secretion, and vesicular transport (see Fig. 1B). It is plausible to assume that these genes were presumably acquired from other bacteria. However, these notions have to be tested. Phyletic patterns as plotted in Table 6 are neutral with respect to this hypothesis.
Environment or lifestyle did not markedly influence the composition of GIs. In both archeal and bacterial GIs, the distributions of COG classes did not significantly differ from the average, superkingdom-specific case. These findings support the notion that HGT serves rather general and perhaps most demanding needs of supplementing metabolic instrumentation. For each superkingdom, selective forces inducing the fixation of HGT events appeared similar, irrespective of the habitats studied here. Due to the concept underlying the COG database, only half of the content of GIs was classified; a mean of 44% of pA genes were annotated as hypothetical (see ratio values in Tables 3 and 4). This fraction is twice as high as for pN genes in nearly all groups. It will be interesting to see which functions are encoded in the remaining genes, whose roles are unclear due to the limitations of current methods in computational biology and where they came from. A putative source of these uncharacterized genes could be unidentified prophages. Recently, it was shown, that viruses possess high genomic diversity (Breitbart et al. 2002). For a precise characterization of these putative genes, it will be necessary to apply biochemical methods. However, it seems plausible to predict and expect mainly operational genes.
References
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T (2003) Informatics for unveiling hidden genome signatures. Genome Res 13:693–702
Berg OG, Kurland CG (2002) Evolution of microbial genomes: sequence acquisition and loss. Mol Biol Evol 19:2265–2276
Borukhov S, Nudler E (2003) RNA polymerase holoenzyme: structure, function and biological implications. Curr Opin Microbiol 6:93–100
Breitbart M, Salomon P, Andresen B, Mahaffy JM, Segall AM, Mead D, Azam F, Rohwer F (2002) Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA 99:14250–14255
Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH (2004) Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci USA 101:3480–3485
Daubin V, Lerat E, Perrière G (2003) The source of laterally transferred genes in bacterial genomes. Genome Biol 4:R57
de la Cruz F, Davies J (2000) Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol 8:128–133
Doolittle WF (1999) Phylogenetic classification and me universal tree. Science 284:2124–2129
Dow JM, Osbourn AE, Wilson TJ, Daniels MJ (1995) A locus determining pathogenicity of Xanthomonas campestris is involved in lipopolysaccharide biosynthesis. Mol Plant Microbe Interact 8:768–777
Eichler J (2003) Facing extremes: archaeal surface-layer (glyco)proteins. Microbiology 149:3347–3351
Florea L, McClelland M, Riemer C, Schwartz S, Miller W (2003) EnteriX 2003: visualization tools for genome alignments of Enterobacteriaceae. Nucleic Acids Res 31:3527–3532
Garcia-Vallvé S, Romeu A, Palau J (2000) Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res 10:1719–1725
Garcia-Vallvé S, Guzman E, Montero MA, Romeu A (2003) HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Res 31:187–189
Genevaux P, Bauda P, DuBow MS, Oudega B (1999) Identification of Tn10 insertions in the rfaG, rfaP, and galU genes involved in lipopolysaccharide core biosynthesis that affect Escherichia coli adhesion. Arch Microbiol 172:1–8
Grantham R, Gautier C, Gouy M, Mercier R, Pave A (1980) Codon catalog usage and the genome hypothesis. Nucleic Acids Res 8:r49–r62
Hacker J, Kaper JB (2000) Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54:641–679
Haft DH, Selengut JD, White O (2003) The TIGRFAMs database of protein families. Nucleic Acids Res 31:371–313
Jain R, Rivera MC, Lake JA (1999) Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA 96:3801–3806
Jeltsch A (2002) Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3:274–293
Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, Zhang X, Zhang J, Yang G, Wu H, Qu D, Dong J, Sun L, Xue Y, Zhao A, Gao Y, Zhu J, Kan B, Ding K, Chen S, Cheng H, Yao Z, He B, Chen R, Ma D, Qiang B, Wen Y, Hou Y, Yu J (2002) Genome sequence of Shegella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res 30:4432–4441
Karlin S (2001) Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol 9:335–343
Karlin S, Mrázek J (2001) Predicted highly expressed and putative alien genes of Deinococcus radiodurans and implications for resistance to ionizing radiation damage. Proc Natl Acad Sci USA 98:5240–5245
Kunst F, Ogasawara N, Moszer I, et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Kurland CG (2000) Something for everyone. Horizontal gene transfer in evolution. EMBO Rep 1:92–95
Kurland CG, Canback B, Berg OG (2003) Horizontal gene transfer: a critical view. Proc Natl Acad Sci USA 100:9658–9662
Lawrence JG, Ochman H (1997) Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol 44:383–397
Lawrence JG, Ochman H (1998) Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA 95:9413–9417
Lawrence JG, Ochman H (2002) Reconciling the many faces of lateral gene transfer. Trends Microbiol 10:1–4
Makarova KS, Koonin EV (2003) Comparative genomics of Archaea: How much have we learned in six years, and what’s next? Genome Biol 4:115
Mantri Y, Williams KP (2004) Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res 32(database issue):D55–D58
Merkl R (2004) SIGI: Score-based identification of genomic islands. BMC Bioinformatics 5:22
Mooi FR, Bik EM (1997) The evolution of epidemic Vibrio cholerae strains. Trends Microbiol 5:161–165
Nakamura Y, Gojobori T, Ikemura T (1999) Codon usage tabulated from the international DNA sequence databases; its status 1999. Nucleic Acids Res 27:292
Nakamura Y, Itoh T, Matsuda H, Gojobori T (2004) Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat Genet 36:760–766
Nesbø CL, Doolittle WF (2003) Targeting clusters of transferred genes in Thermotoga maritima. Environ Microbiol 5:1144–1154
Nesbø CL, L’Haridon S, Stetter KO, Doolittle WF (2001) Phylogenetic analyses of two “archaeal” genes in Thermotoga maritima reveal multiple transfers between Archaea and Bacteria. Mol Biol Evol 18:362–375
Nicolas P, Bize L, Muri F, Hoebeke M, Rodolphe F, Ehrlich SD, Prum B, Bessières P (2002) Mining Bacillus subtilis chromosome heterogeneities using hidden Markov models. Nucleic Acids Res 30:1418–1426
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304
Ohnishi M, Kurokawa K, Hayashi T (2001) Diversification of Escherichia coli genomes: are bacteriophages the major contributors? Trends Microbiol 9:481–485
Raetz CR, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700
Ragan MA (2001a) Detection of lateral gene transfer among microbial genomes. Curr Opin Genet Dev 11:620–626
Ragan MA (2001b) On surrogate methods for detecting lateral gene transfer. FEMS Microbiol Lett 201:187–191
Raymond CK, Sims EH, Kas A, Spencer DH, Kutyavin TV, Ivey RG, Zhou Y, Kaul R, Clendenning JB, Olson MV (2002) Genetic variation at the O-antigen biosynthetic locus in Pseudpmanas aeruginosa. J Bacteriol 184:3614–3622
Ruepp A, Graml W, Santos-Martinez ML, Koretke KK, Volker C, Mewes HW, Frishman D, Stocker S, Lupas AN, Baumeister W (2000) The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407:508–513
Tatusov RL, Koonin EV, Lipman DJ (1997) A genomic perspective on protein families. Science 278:631–637
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA (2003) The COG database: an updated version includes Eukaryotes. BMC Bioinform 4:41
Tu Q, Ding D (2003) Detecting pathogenicity islands and anomalous gene clusters by iterative discriminant analysis. FEMS Microbiol Lett 221:262–275
Wang HC, Badger J, Kearney P, Li M (2001) Analysis of codon usage patterns of bacterial genomes using the self-organizing map. Mol Biol Evol 18:792–800
Acknowledgments
This project was carried out within the framework of the Competence Network Göttingen “Genome Research on Bacteria” (BiotechGenoMik) financed by the German Federal Ministry of Education and Research (BMBF). I thank Rüdiger Schmidt for his help in preparing the manuscript and the referees for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Siv Andersson]
Rights and permissions
About this article
Cite this article
Merkl, R. A Comparative Categorization of Protein Function Encoded in Bacterial or Archeal Genomic Islands. J Mol Evol 62, 1–14 (2006). https://doi.org/10.1007/s00239-004-0311-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0311-5