
Abstract
In this study the molecular evolution of duplicated HoxA genes in zebrafish and fugu has been investigated. All 18 duplicated HoxA genes studied have a higher non-synonymous substitution rate than the corresponding genes in either bichir or paddlefish, where these genes are not duplicated. The higher rate of evolution is not due solely to a higher non-synonymous-to-synonymous rate ratio but to an increase in both the non-synonymous as well as the synonymous substitution rate. The synonymous rate increase can be explained by a change in base composition, codon usage, or mutation rate. We found no changes in nucleotide composition or codon bias. Thus, we suggest that the HoxA genes may experience an increased mutation rate following cluster duplication. In the non-Hox nuclear gene RAG1 only an increase in non-synonymous substitutions could be detected, suggesting that the increased mutation rate is specific to duplicated Hox clusters and might be related to the structural instability of Hox clusters following duplication. The divergence among paralog genes tends to be asymmetric, with one paralog diverging faster than the other. In fugu, all b-paralogs diverge faster than the a-paralogs, while in zebrafish Hoxa-13a diverges faster. This asymmetry corresponds to the asymmetry in the divergence rate of conserved non-coding sequences, i.e., putative cis-regulatory elements. These results suggest that the 5′ HoxA genes in the same cluster belong to a co-evolutionary unit in which genes have a tendency to diverge together.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hox cluster duplications play a prominent but poorly understood role in vertebrate evolution (Wagner et al. 2003). Hox genes code for homeodomain containing transcription factors and are homologous to the homeotic genes in the Drosophila Antp and Ubx gene clusters. The Hox gene family is thought to have arisen by tandem duplications, leading to a cluster of three or four homeobox containing genes, which was then duplicated, giving rise to the ancestral Hox and para-Hox gene clusters (Holland and Garcia-Fernandez 1996; Kappen et al. 1989). Additional tandem duplications increased the number of Hox genes in the cluster to up to 14 in chordates (Ferrier et al. 2000; Powers and Amemiya 2004). All well-characterized invertebrate taxa have a single cluster with the exception of the nematode, Caenorhabditis elegans, and the urochordates, Ciona intestinalis and Oikopleura dioica, which have lost the integrity of the Hox cluster all together (Dehal and et al. 2002; Ruvkun and Hobert 1998; Seo et al. 2004). In contrast, multiple copies of Hox clusters have been found in all vertebrate lineages, varying from at least three in lamprey to seven or eight in teleost species (Holland et al. 1994; Martinez and Amemiya 2002; Meyer and Malaga-Trillo 1999; Prohaska and Stadler 2004; Ruddle et al. 1994b; Taylor et al. 2003). From these data at least three duplication events can be inferred, although the number of duplications might be as high as seven (Stadler et al. 2004). The reason for the different pattern of Hox cluster evolution in vertebrate compared to invertebrate lineages is not clear. One possibility is that the duplicated genes are necessary for vertebrate body plan elaboration (Holland et al. 1994; Malaga-Trillo and Meyer 2001; Ruddle et al. 1994a) or to overcome structural constraints specific to vertebrate Hox clusters (Fried et al. 2004; Wagner et al. 2003).
A variety of teleost species is known to have more than four Hox clusters (Meyer and Schartl 1999; Prince and Pickett 2002; Prohaska and Stadler 2004), including the zebrafish (Amores et al. 1998), fugu and Spheroides (Amores et al. 2004), and medaka (Naruse et al. 2000, 2004). Preliminary data also exist for the killifish (Misof and Wagner 1996), striped bass (Pavell and Stellwag 1994; Snell et al. 1999), tilapia (Santini et al. 2003), and rice field eel (Ji et al. 2002). It is not entirely clear when this duplication has happened, but data suggest that it was some time after the most recent common ancestor of paddlefish and other ray fined fishes and before the most recent common ancestor of Euteleosts (Metscher et al. 2005).
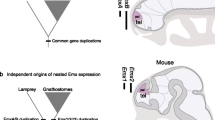
In this paper, we examine the evolutionary fate of duplicated HoxA genes in the teleosts in order to gain insight into the role Hox cluster duplication plays in vertebrate evolution. Three sets of taxa are considered in this paper, one for which Hox gene sequences are available and one where RAG1 sequence information is known and the non-coding sequences of zebrafish, fugu, tilapia, bichir, and horn shark HoxA clusters. The phylogenetic hypotheses for these two sets of species are given in Figs. 1A and 1B.

Phylogenetic relationships among the gene sequences analyzed in this paper. A Gene tree of the HoxA genes analyzed. Bichir is considered the most basal ray finffed fish lineage. Paddlefish together with the sturgeons belongs to the second most basal ray finned fish lineage (Inoue et al. 2003), and the two paralogs of the HoxA clusters in fugu and zebrafish derive from the same duplication event (Amores et al. 2004). B Phylogenetic relationships among the RAG1 genes analyzed in this paper. The bowfin, Amia calva, and paddlefish are considered to belong to a clade according to the results of Inoue et al. (2003). The relationships of the osteoglossomorph sequences (Osteroglossus, Panthodon. Gnathonbemus, and Notopterus) are based on Guo-Qing and Wilson (1996).
Materials and Methods
Data
Full Hox cluster sequences were used in this study from the shark, Heterodontus francisci (HfHoA = AF479755); the bichir, Polypterus senegalus (PsHoxA = AC132195 and AC12632); the two zebrafish paralogs, Danio rerio (DrHoxAa = AC107365 r.c. and DrHoxAb = AC107364); the fugu, Takifugu rubripes (TrHoxAa = Fugu v.3.0 scaffold 47, positions 103,001–223,000 r.c., contains FRU92573; and TrHoxAb = Fugu v.2.0 scaffold 1874); and tilapia, Oreochromis niloticus (OnHoxA = AF533976).
The Hoxa-13 and Hoxa-11 sequences of paddlefish were PCR amplified, cloned and sequenced, and described elsewhere (Metscher et al. 2004). The sequences are published In GenBank (Hoxa-13: AY661749, protein ID AAT75331) (Hoxa-11: genomic sequence AY661748, protein ID AAT75330).
RAG1 sequences of the following species were retrieved from GenBank: zebrafish, Danio rerio (NM_131389); fugu, Takifugu rubripes (AF108420); winter flounder, Pseudopleuronectus americanus (AF369067); Atlantic cod, Gadus morhua (AF369064); Osteoglossomorpha: bronze beatherback, Notopterus notopterus (AF369063), Gnathonemus sp. (IMCB-2001) (AF369062), freshwater butterfly fish, Pantodon buchholzi (AF369061), Osteoglossum sp. (AF369060); bowfin, Amia calva (AF369059); Mississippi paddlefish, Polyodon spathula (AF369057); bichir, Polypterus sp. (IMCB-2001) (AF369055); and the coelacanth, Latimeria menadoensis (AF369069).
Analysis of Coding Sequence Evolution
Both the nucleotide sequence and predicted amino acid Sequence were aligned with ClustalW. The amino acid sequence was then inspected for regions of ambiguous alignment. These regions and gaps were eliminated from the alignment. Finally, the nucleotide sequence corrected to conform to the edited amino acid sequence. Sequence alignments were analyzed with three programs as appropriate and indicated in the Results: RRTree 1.1.11 (Robinson-Rechavi and Huchon 2000), HYPHY99.beta for MacOS (Muse and Gaut 1994), and PAML (Yang 1997).
Relative Rate Test for Conserved Non-CodingSequences
Conserved non-coding sequences were detected using the tracker program (Prohaska et al. 2004). Very briefly, this approach is based on BLAST (Altschul et al. 1990) for the initial search of all pairs of input sequences restricted to homologous intergenic regions. The resulting list of pairwise sequence alignments is then assembled into groups of partially overlapping regions that are subsequently passed through several filtering steps and finally aligned using the segment based multiple alignment tool DIALIGN2 (Morgenstern 1999). The final output of the program is the list of these aligned “footprint cliques” (see supplemental material at http://www.tbi.univie.ac.at/Publications/SUPPLEMENTS/04-014). The alignments of all footprint cliques are concatenated and padded with gap characters where a footprint detected between some sequences does not have a counterpart in others. Consequently, all gap characters are treated as unknown nucleotides rather than as deletions. Conserved sequences between two outgroup species, in this case the shark, Heterodontus francisci, and the bichir, Polypterus senegalus, are compared to pairs of ingroup sequences, in these cases all pairs of the zebrafish a/b clusters, the fugu a/b clusters, and the tilapia, Oreochromis niloticus, HoxAa cluster. The fraction of the conserved nucleotide positions between the two ingroup sequences is compared with a exponential decay model (Wagner et al. 2004), assuming that there is a constant rate of loss of conservation along the lineages. Finally, these numbers are used for testing for differences in the rate of modification between the two ingroup lineages as described in Wagner et al. (2004).
Background
Hox cluster duplication is followed by loss of some duplicated genes. The gene complement of various ray finned fish HoxA clusters has been described in previous papers (bichir [Chiu et al. 2004]; zebrafish [Amores et al. 1998; Chiu et al. 2002]; stripped bass [Snell et al. 1999]; fugu and puffer fish [Amores et al. 2004]; tilapia [Santini et al. 2003]). In addition, the complete sequence of the horn shark HoxA cluster has been described (Chiu et al. 2002; Kim et al. 2000). Here we recapitulate the salient features of these results to set the stage for the detailed sequence analysis reported below (Fig. 2).

The gene inventory of the HoxA clusters analyzed in this paper. The shark, Heteroaontus francisci, HoxA cluster, HfHoxA, has been described in Kim et al. (2000) and Chiu et al. (2002) as the HoxM cluster. The coelacanth. Latimeria menadoensis, HoxA cluster, LmHoxA, is described in Powers et al. (2004), and the bichir, Polypterus senegalus, cluster, PsHoxA, has been described in Chiu et al. (2004). The paddlefish Hox genes are only known from three partial HoxA gene sequences: Hoxa-13 and Hoxa-11 are represented by almost-complete coding sequences including the intron (Metscher et al. 2004)and Hoxa-1 by a smaller fragment (J. McClintock, C. Jozefowicz, and V.E. Prince; AY 188522). The zebrafish HoxA clusters, DrHoxAa/b, are based on the complete Hox cluster sequence described in Chiu et al. (2002), and the fugu sequences, TrHoxAa/b, are from the fugu genome sequence versions 2.0 and 3.
The HoxA cluster of the ancestral ray finned fish most likely had paralog group members 1 to 7, 9 to 11, and 13 and 14, i.e., 12 genes (Powers and Amemiya 2004). Prior to HoxA cluster duplication in the stem of teleost fishes the HoxA cluster further lost Hoxa-6 and Hoxa-14 (Chiu et al. 2004; Powers and Amemiya 2004), and thus had a total of 10 genes. After duplication the b-paralog cluster lost all the anterior and medial genes with the exception of Hoxa-2b. In contrast the HoxAa paralog cluster only lost one or two genes in different lineages. The fugu and pufferfish lineage both lost Hoxa-7a and zebrafish also Hoxa-10a, while tilapia and the striped bass retained both of these genes. The fugu has the largest number of first order paralogs, namely ,all the AbdB related genes Hoxa-9a/b, Hoxa-10a/b, Hoxa-11a/b, and Hoxa-13a/b, as well as Hoxa-2a/b. In zebrafish only three 1st order paralog HoxA genes are maintained: Hoxa-9 a/b, Hoxa-11a/b, and Hoxa-13a/b. The detailed sequence analysis presented here focuses on the Hox genes with 1-order paralogs, with particular focus on Hoxa-11 and Hoxa-13, and some results for Hoxa-2, Hoxa-9, and Hoxa-10. The analysis of the divergence of non-coding sequences of fugu and zebrafish will be restricted to the intergenic regions between Hoxa-9 and Hoxa-13, i.e., the segment of the cluster that contains most of the 1st-order paralogs.
Inferences on the consequences of Hox gene duplication depend critically on the availability of close outgroup taxa. There are only limited data from basal actinopterygian HoxA clusters. Recently we described a complete HoxA cluster sequence from the most basal lineage of ray finned fishes, the bichir (Chiu et al. 2004), and two genes, Hoxa-11 and Hoxa-13, from the American paddlefish, Polyodon spathula (Metscher et al. 2005), which is a member of the second most basal ray finned fish lineage (Bermis et al. 1997). Gene tree analysis of either taxon shows that these lineages arose prior to the origin of the paralog HoxA clusters found in the teleosts (Chiu et al. 2004; Metscher et al. 2005).
Results
Increased Rate of Synonymous and Non-Synonymous Substitutions After Hox Gene duplication
The amino acid substitution rates of the 18 teleost Hox genes studied here were compared to the rate in either the paddlefish or the bichir using a maximum likelihood relative rate test as implemented in HYPHY99beta for MacOS. In every instance the estimated amino acid substitution rate in the teleost gene was higher than that in bichir or paddlefish and in 15 cases the difference was statistically significant (Fig. 3, Table 1).

Unambiguously reconstructed non-synonymous substitutions of Hoxa-11 (A) and Hoxa-13 (B) mapped onto the gene tree by maximum parsimony. The tree is on the phylogenetic tree of the involved species and the fact that the duplicated genes in zebrafish and fugu are orthologous (Amores et al. 2004). Note that the duplicated genes are accelerated compared to the non-duplicated genes of paddlefish, bichir, and coelacanth.
The higher rate of non-synonymous substitutions in duplicated Hox genes is expected assuming that duplicated genes have a certain degree of functional redundancy. Functional redundancy would imply that the non-synonymous rate would differentially increase over the synonymous rates, leading to an increased dN/dS ratio, ω. Given that the duplication may be as long as 320 mya (Vanderpoele et al. 2004), estimating the synonymous substitution rate is only possible with genes for which relatively close outgroups are available. Table 2 gives the results for Hoxa-11 and Hoxa-13, since only for those genes did we have a paddlefish ortholog at hand. Both dN and dS were estimated with a codon based maximum likelihood method (Codeml of PAML) (Yang 1997) and a distance based method (RRTree) (Robinson-Rechavi and Huchon 2000) and compared to those in paddlefish (Tables 2A and B). Both methods account for phylogenetic structure, which makes these methods more appropriate for the comparison of deeply divided lineages than the three taxa relative-rate tests. In addition, RRTree estimates the transversion rate at fourfold degenerate sites (B4). This measure of the synonymous substitution rate can be estimated over longer phylogenetic distances because it saturates more slowly than dS itself.
PAML-based tests for differences in ω between teleosts and paddlefish did not show significant differences (not shown) in spite of the large differences in amino acid substitution rates summarized in Table 1. The data in Tables 2A and B show that both the synonymous and the non-synonymous substitution rate in the teleost lineage are increased by a factor of 3 to 5, depending on the gene and the method of estimation. For Hoxa-11 both dS and B4 are significantly higher in teleosts than in paddlefish according to RRTree. For Hoxa-13 dS could not be estimated but B4 is significantly increased (differences in dS cannot be tested in PAML). The increased synonymous substitution rate could be the result of changes in the base composition and/or codon bias or an increased mutation rate. Below we show that the first two explanations are not supported by the data.
Accelerated Evolution After Hox Cluster Duplication Is Not Caused By Changes in Composition or Codon Bias
To address the issue why the rate of synonymous substitutions increased after Hox cluster duplications, we consider three possibilities: a systematic shift in the base composition of the HoxA clusters, a change in codon bias, and a general increase in mutation rate in all nuclear genes.
As shown in Tables 2A and B, the GC content of the teleost HoxA clusters and genes does not differ systematically from outgroups. On average there is a slightly higher GC content of teleost genes but this difference is only due to perciform species, i.e., fugu, tiliapia,and sliped bass, and not zebrafish (data not shown), which also has an increased dS. Hence there is no indication that the change in synonymous substitution rate is due to a shift in GC content. Similarly, the effective number of codons, Nc, a measure of codon bias (Wright 1990), is not systematically different between teleosts and outgroups like bichir, coelacanth, shark, or paddlefish. There are a few genes which have changed their codon usage pattern, but they are single genes in single lineages, like fugu Hoxa-2b, and cannot account for the overall increase in synonymous substitution rate. Thus we conclude that the most likely explanation for the increased synonymous substitution rate is an increase in mutation rate.
In order to test whether the apparent increase in mutation rate is limited to HoxA genes or a genomewide pattern, we examined another nuclear gene, RAG1. As mentioned above, the main limitation for detecting any rate differences in sequence evolution is the availability of close outgroup taxa. We only found one gene with sufficient coverage of basal ray finned fish lineages in GenBank to estimate the synonymous rate differences between teleosts and basal ray finned fishes, RAG1.
Rate of RAG1 Evolution in Ray Finned Fishes
We obtained RAG1 sequences from 12 species to compare the rate of evolution between teleost and basal ray finned fish lineages, in particular, paddlefish and Amia. The rate of evolution was estimated and tested for difference with three methods: codon based PAML (Yang 1997), codon model of Muse and Gaut (1994) as implemented in HYPHY.99beta, and tree based relative rate tests as implemented in RRTree (Robinson-Rechavi and Huchon 2000). RRTree tests for differences in nonsynonymous substitution rates are all highly significant between Amia/paddlefish and teleosts, as well as between Amia/paddlefish and osteoglossomorphs. Hence teleost nuclear genes investigated here evolve at a higher non-synonymous rate than close outgroups.
The synonymous substitution rate could not be estimated with RRTree due to saturation and even B4, the fourfold degenerate transversion rate, could only be estimated with exceedingly high standard deviations. We then performed an RRTree test with the 3rd codon positions. The nucleotide substitution rate at 3rd positions could be estimated with reasonable error variance and indicated no difference between teleost clades and Amia/paddlefish. The synonymous substitution rates estimated by PAML are consistently higher in teleosts but no direct test for the significance of these estimates can be given in the PAML framework.
Finally, we performed a local clock test based on the Muse and Gaut (1994) codon model for heterogeneity of dS rates. The results show that the teleosts are not different from a clock model, and neither is a group including the Amia and paddlefish. Hence synonymous substitution rate is not distinguishable from a clock model in a clade which includes the basal rayfinned fishes Amia and paddlefish as well as teleosts. A local clock test for non-synonymous substitutions, however, provides strong evidence for rate heterogeneity within this clade (LR=48.52, P=810−8) but not within the teleosts clade. This is consistent with the results of tree based relative rate tests (see above).
In order to test the power of the local clock model to detect differences in the synonymous substitution rate, we performed a local clock test with the Hoxa-11 data. The results indicate significant rate heterogeneity at the level of inclusion predicted by the RRTree tests, i.e., significant heterogeneity in the clade including paddlefish but not within the teleost gene clade. Hence the clock test for synonymous rates is powerful enough to detect synonymous rate heterogeneity in Hoxa-11 sequences at that level of phylogenetic differentiation and with shorter sequences than the RAG1 alignment. Hence it seems likely that the failure of detecting synonymous rate heterogeneity for RAG1 among the ray finned fishes is not due to lack of power. It rather seems that there is a much smaller synonymous substitution rate difference than found in the Hox genes, between teleosts on one side and Amia and paddlefish on the other side.
In summary, there is strong evidence for an increased non-synonymous substitution rate in teleosts for RAG1, but no evidence for an increase in the synonymous substitution rate in teleosts. While there are still questions about the power to detect differences in the synonymous substitution rate with taxa as diverged as paddlefish and zebrafish, the available results from local clock tests and the rate of 3rd codon positions argues against that possibility. Hence it is possible that the increased dS rate observed for HoxA genes is Hox gene specific rather than a genomewide phenomenon.
Asymmetry of Divergence Among 1st-Order Paralogs
Among the zebrafish HoxA genes there are three 1st-order paralog gene pairs: Hoxa-13a/b, Hoxa-11a/b and Hoxa-9a/b (Fig. 2). Among those genes only one, Hoxa-13a, has a significantly increased non-synonymous substitution rate compared to its 1st-order paralog Hoxa-13b (Table 3). Fugu, on the other hand, has five 1st-order HoxA paralog pairs: Hoxa-13a/b, Hoxa-11a/b, Hoxa-10a/b, Hoxa-9a/b, and Hoxa-2a/b (Fig. 2). Of these, three have a significantly increased rate of non-synonymous substitutions, namely, Hoxa-11b, Hoxa-10b, and Hoxa-2b (Table 3). Note that all of the accelerated genes are b-paralogs. Furthermore, the estimated rates for all of the b-paralogs are higher than those of the a-paralogs even for those two cases where the differences are not statistically significant, Hoxa-13a/b and Hoxa-9a/b. The a priori probability that in five instances paralogs from the same cluster have a higher estimated rate than the other is 0.55 = 0.03125, <5%. Hence it is possible that in fugu the b-paralogs are diverging at a systematically higher rate than the a-paralogs, rather than paralogs randomly sampled from both paralog clusters.
Asymmetry of Divergence of Conserved Non-codins Sequences
The majority of 1st-order paralog gene pairs are found in the 5′ segment of the HoxA gene cluster. To compare the rate of non-coding sequence evolution after cluster duplication, we focused on the inter-genie sequences 3′ of Hoxa-13 and 5′ of Hoxa-9. In this analysis we also included the published sequence of the Tilapia HoxA-a cluster (Santini et al. 2003). A global alignment of the 5′HoxA clusters of shark and bichir identified blocks of conserved nucleotides as described in Prohaska et al. (2004). Then we identified the corresponding blocks of conservation in the five teleost HoxA clusters from zebrafish, fugu, and tilapia and subjected them to a relative rate test as described in (Wagner et al. 2004). Three of the duplicated 5′HoxA clusters are indistinguishable with respect to the rate of modification of conserved non-coding sequences among the clusters: the zebrafish HoxA-b, tilapia HoxA-a, and fugu HoxA-a clusters (Table 4). The zebrafish Hox cluster segment DrAa13-9, i.e., the a-paralog, retains significantly fewer conserved nucleotide positions (about 20%) than its zebrafish paralog, DrAb13-9, as well its orthologs in fugu and tilapia, TrAal3-9 and OnAa13-9. The fugu TrAbl3-9, the b-paralog, evolves faster in its putative cis-regulatory elements than its paralog TrAa13-9 and its zebrafish ortholog DrAb13-9. Two clusters show a significant increase in the rate of modification of conserved non-coding sequences— zebrafish HoxA-a and fugu HoxA-b clusters—but their rates of divergence are indistinguishable from each other.
The results of the asymmetric modification of conserved non-coding sequences show an intriguing pattern of congruence with the divergence of the coding sequences. In zebrafish the only gene with asymmetric divergence is a HoxA-a paralog, Hoxa-13a, and the non-coding sequences of the HoxA-a cluster are diverging at a higher rate than that of the zebrafish HoxA-b cluster. There is however, a possible confounding factor in this comparison since it is also the HoxA-a cluster that lost its Hoxa-10a paralog and thus may have lost more conserved non-coding sequence than HoxA-b cluster.
In fugu, however, the pattern is clearer. All five b-paralogs are estimated to have increased rates of non-synonymous substitutions (although only three of them are statistically significant; Table 3), and also the non-coding sequences of the HoxA-b cluster diverge faster than the fugu HoxA-a cluster. Notably the non-coding sequences of the fugu HoxA-b cluster also diverge faster than the zebrafish HoxA-b cluster. The latter observation is significant because both the zebrafish and the fugu HoxA-b clusters have the same gene inventory (Fig. 2). Hence the increased rate of non-coding sequence evolution in the fugu HoxA-b cluster cannot be explained by differences in the number of genes retained. Thus in the fugu we observe a concerted asymmetric divergence of both the coding as well as the non-coding sequences of the 5′HoxA-b clusters after duplication. Furthermore, this pattern is distinctly different from that observed in the zebrafish, where the HoxA-a cluster divergences at a higher rate. Hence the majority of the asymmetric divergence occurred after the split of the zebrafish and the fugu lineage, rather than immediately after Hox cluster duplication.
In order to examine whether the non-coding sequence evolution rate increase is uniform along the Hox cluster, we performed relative rate tests on each of the intergenic regions independently (Fig. 4). In the zebrafish HoxAa cluster the higher rate of divergence of the a-paralog is focused on the intergenic regions between Hoxa-13 and Hoxa-11 as well as between Hoxa-11 and Hoxa-10. These two regions are 3′ of Hoxa-13, which has an accelerated rate of non-synonymous divergence, and 5′ of Hoxa-10, which in the HoxAa cluster is a pseudogene. In fugu, the only significantly increased rate of non-coding divergence is between Hoxa-10 and Hoxa-9, where Hoxa-10 has an accelerated rate of evolution. Hence, the rate acceleration is differential among different ingergenic regions and could provide hints at the molecular level of regulatory changes following Hox cluster duplications.

Spatial distribution of sequence divergence along the 5′ segment of the duplicated teleost HoxA clusters. The open boxes symbolize the coding regions (ignoring introns), and the lines the intergenic sequences (not to scale). The gray boxes in the intergenic regions give the difference in the fraction of conserved non-coding sequence positions modified in one of the two paralogs: (1−r(HoxAa))−(1-r(HoxAb)). If the difference is >0, i.e., if the HoxAa cluster is more modified than the HoxAb cluster, the bar is on top of the line; if the HoxAb is more modified, the bar is drawn below the line. In the coding regions the gray bars symbolize the difference in the rate of non-synonymous substitutions (Table 3). *p < 5%, **p < 2.5%, ***p < 1%; n.s., p < 5%.
Discussion
Rate of Sequence Evolution in Teleosts
For all genes analyzed in this paper, including the non-Hox gene RAG1, the rate of non-synonymous substitutions is higher in teleosts than in close out groups, i.e., basal bony fish lineages like paddlefish and bichir. This finding is consistent with at least two previously published reports. Robinson-Rechavi and Laudet (2001) reported that most nuclear genes evolve quicker in teleosts than in mammals, independent of the presence of paralog genes, i.e., teleost genes evolve quicker regardless of whether there is potential for genetic redundancy or not. Krieger and Fuerst (2002) reported a lower rate of amino acid evolution in sturgeons compared to teleost genes, in accordance to the results reported here. The interpretation of these authors, however, was that the rate in sturgeons is depressed, but this feature is shared with all basal gnathostome lineages, including bichir and Latimeria. Hence it is more parsimonious to assume that the high rate of amino acid sequence evolution in teleosts is derived, rather than the low rate in bichir and paddlefish.
In teleost Hox genes the increased rate of non-synonymous substitutions is not necessarily indicative of relaxed purifying selection or directional selection. Other studies have established that recently duplicated genes have a significantly increased dN/dS rate ratio ω, indicating either relaxed selection or directional selection (Conant and Wagner 2003; Kondrashov et al. 2002; Lynch and Conery 2000). The failure to detect an increased ω in teleost Hox genes may be due to two factors. First, the duplication of teleost genes is likely associated with a genome duplication which may have occurred as long as 320 mya (Vanderpoele et al. 2004). Lynch and Connery (2000) have shown that older duplicates return to lower ω values than recent duplicates. On the other hand, we report evidence that the synonymous substitution rate, dS, is also substantially increased. An increased rate of dS makes it harder to detect relaxed stabilizing selection.
Not many studies have addressed the questions whether synonymous substitution rates differ between teleosts and other bony fish. Merrit and Quattro studied the evolution of cytosolic malate dehydrogenase genes and reported an increased rate of evolution of teleost genes compared to sturgeon and mammalian genes (Merrit and Quattro 2003). The authors report an increased dN/dS ratio and even directional selection in one lineage, i.e., dN/dS>1. The average number of synonymous changes in the teleost lineages (111.75), however, is similar to that in the sturgeon lineage (99) and not comparable with the roughly threefold increase we found in the HoxA genes in this study. We are not aware of any study that has investigated dS for other teleost nuclear genes and compared it with basal actinopterygians. Our attempts to test whether the non-Hox gene RAG1 also has increased dS were ambiguous, but the balance of evidence suggests that dS for RAG1 is not increased among teleosts, at least not as much as in HoxA genes. Hence, it is possible that the increased rate of synonymous substitutions is a phenomenon restricted to duplicated Hox clusters or may be even only the HoxA clusters of teleosts.
Of the possible explanations for an increased rate of synonymous substitutions, we could eliminate two: changes in base composition and changes in the degree of codon bias. We thus conclude that the most likely explanation for the increased dS is an increased mutation rate but also note that this conclusion is supported only by the elimination of known alternatives and not by direct evidence. It would be desirable to compare the mutation rates of homologous genes in teleost and non-teletost fishes.
Asymmetrical Divergence of Paralog Genes
There are a total of eight 1st-order HoxA paralog gene pairs, three gene pairs in zebrafish and five in fugu. Four of those paralog pairs (50%) diverge at a rate that is statistically distinguishable at the 5% level. This fraction is comparable of that reported by Zhang and collaborators (2003) for recent duplicates in the human genome (60%). Conant and Wagner (2003) report asymmetry in 20 to 30% of duplicated yeast, Drosophila, and Caenohabditis elegans genomes. Van de Peer and collaborators (2001) found that 50% of duplicated zebrafish genes have significant asymmetry in divergence. Kellis et al. (2004) report about 16% asymmetrically diverging genes in yeast, although these numbers are not comparable because they did not apply rigorous statistical tests to detect asymmetry. In contrast, Kondrashov et al. (2002) only found significant asymmetry in 2 of 49 (4%) recently duplicated mammalian genes and 2 of 11 (18%) in Drosophila. While there might be both biological as well as methodological reasons responsible for these discrepancies, the results show that the HoxA genes in zebrafish and fugu are at the upper end of the frequency of asymmetrically diverging genes families even though they are not recently duplicated like the mammalian genes reported by Zhang et al. (2003). This result is consistent with that of Van de Peer et al. (Peer et al. 2001), who also found high levels of asymmetrical divergence in a sample of genes containing a large fraction of transcription factors.
In fugu the asymmetrical divergence of duplicated HoxA genes has two intriguing additional features. In all of the five 1st-order paralog HoxA gene pairs, the b-paralogs have a higher estimated rate of dN than the a-paralog (of which three are statistically significant), which by chance is less likely than 5%. It is thus possible that the asymmetric divergence of Hox genes on a cluster is not independent but is correlated perhaps because of the strong regulatory integration among the Hox genes. Acquisition of new functions by one Hox gene may increase the chance of functional recruitment of other genes on the same cluster. This conclusion is also supported by the fact that many secondary functions of AbdB Hox genes, like their role in paired appendage development and reproductive tract development, involve multiple Hox genes and, often, all the AbdB related genes on a cluster (Haack and Gruss 1993; Kobayashi and Behringer 2003; Taylor et al. 1997). These facts suggest that the set of AbdB related genes acts like a co-evolutionary module (Schlosser 2002).
Furthermore, the conserved non-coding regions in the cluster segment 5′ of Hoxa-9 (the AbdB related set of genes) also diverge asymmetrically, with the fugu 5′HoxA-b cluster evolving faster. This suggests that in fugu the HoxA-b cluster diverged faster with respect to protein coding sequences as well as putative cis-regulatory elements. In the zebrafish the HoxA-b cluster is neither accelerated in any of its protein coding genes nor with respect to its non-coding sequences. The situation in the zebrafish HoxA-a cluster is complicated due to a higher rate of gene loss than the fugu HoxA-a cluster. In any case the results show that the pattern of asymmetric gene divergence differs between the zebrafish and the fugu lineage.
In both the zebrafish HoxAa cluster and the fugu HoxAb cluster the rate acceleration of non-coding sequence evolution is heterogeneous along the cluster. In either case the intergenic segments that show accelerated evolution are localized in the vicinity of coding regions which diverge asymmetrically or the site of recent pseudogenes (Fig. 4). Thus, it is likely that the regulatory changes that lead to the asymmetrical divergence of Hox genes are localized and thus specific to particular sequence domains rather than distributed general along the whole paralog sequences.
The relationship between the rate of cis-regulatory element evolution and the rate of protein evolution has recently been investigated in nematodes (Castillo-Davis et al. 2004). These authors found no significant correlation between dN and the divergence of promoter proximal transcription factor binding sites if the regression is corrected for differences in dS, i.e., time of duplication and mutation rate. Hence the association reported here between putative cis-regulatory element evolution and Hox protein divergence was not found in a genomewide study. The reason for this discrepancy might be that the mechanistic causes for protein divergence differs between different classes of proteins. Castillio-Davis and collaborators did not report whether the correlation between cis-regulatory element divergence and protein sequence divergence differs between different classes of proteins. For transcription factors differences in tissue expression are possibly associated with a different set of tissue specific protein interaction partners (Featherstone 2003). Hence it is mechanistically plausible that divergence in gene regulation, inferred from the divergence in putative cis-regulatory elements, and protein sequence evolution are more strongly correlated in transcription factor genes than in other genes.
It is noteworthy that the asymmetric divergence of paralog genes is different between the zebrafish and the fugu lineage. For instance, in zebrafish the Hoxa-13a paralog is diverging faster, while in fugu it is the Hoxa-13b paralog. This implies that the faster divergence happened after the split of the zebrafish and fugu lineages rather than immediately after the duplication event, suggesting that asymmetrical divergence is driven by forces that do not acting immediately after the duplication but take some time to mount and are random among lineages. This result could also explain why the frequency of asymmetrically diverging duplicated genes is very low among recently duplicated genes (4 to 18% [Kondrashov et al. 2002]). An interpretation of this pattern is that asymmetrical divergence is indicative of a novel function by the faster diverging gene (this possibility is considered further below with the zebrafish Hoxa-13 genes). The acquisition of a novel function is probably only possible after a considerable period of random divergence due to relaxed stabilizing selection. This scenario would explain the delay after duplication in the development of asymmetric divergence.
Conclusions
Based on the results reported here we conclude that there are two stages of Hox gene evolution following Hox cluster duplication, consistent with the idea that duplicated Hox genes can contribute to physiological or developmental innovations (Malaga-Trillo and Meyer 2001; Ohno 1970; Wagner et al. 2003).
-
1
Immediately following the Hox cluster duplication there was a phase of increased random divergence including gene loss and increased mutation rate.
-
2
Some time after the duplication one paralog starts to diverge faster than the other paralog.
References
SF Altschul W Gish W Miller EW Myers DJ Lipman (1990) ArticleTitleBasic local alignment search tool J Mol Biol 215 403–410 Occurrence Handle10.1006/jmbi.1990.9999 Occurrence Handle1:CAS:528:DyaK3MXitVGmsA%3D%3D Occurrence Handle2231712
A Amores A Force Y-L Yan L Joly C Amemiya A Fritz RK Ho J Langeland V Prince Y-L Wang M Westerfield M Ekker HH Postlethwait (1998) ArticleTitleZebrafish hox clusters and vertebrate evolution Science 282 1711–1714 Occurrence Handle1:CAS:528:DyaK1cXnslGgtrY%3D Occurrence Handle9831563
A Amores T Suzuki Y-L Yan J Pomroy A Singer C Amemiya J Postlethwait (2004) ArticleTitleDevelopmental roles of pufferfish Hox clusters and genome evolution in ray-fin fish Genome Res 14 1–10 Occurrence Handle1:CAS:528:DC%2BD2cXlvVGntg%3D%3D Occurrence Handle14707165
WE Bermis EK Findeis L Grande (1997) ArticleTitleAn overview of Acipenseriformes Environ Biol Fish 48 25–71
CI Castillo-Davis FA Kondrashov DL Hartl RJ Kulathinal (2004) ArticleTitleThe functional genomic distribution of protein divergence in two animal phyla: coevolution, genomic conflict, and constraint Genome Res 14 802–811 Occurrence Handle1:CAS:528:DC%2BD2cXjvFyktbs%3D Occurrence Handle15123580
Chiu C-h C Amemiya K Dewar Kim C-b F Ruddle GP Wagner (2002) ArticleTitleMolecular evolution of the HoxA cluster in three major gnathostome lineages Proc Natl Acad Sci USA 99 5492–5497 Occurrence Handle10.1073/pnas.052709899 Occurrence Handle1:CAS:528:DC%2BD38XjtFKltrw%3D Occurrence Handle11943847
Chiu C-h K Dewar GP Wagner K Takahashi F Ruddle C Ledje P Bartsch J-L Scemama E Stellwag C Fried SJ Prohaska PF Stadler CT Amemiya (2004) ArticleTitleBichir HoxA cluster sequence reveals surprising trends in ray-finned fish genomic evolution Genome Res 14 11–17 Occurrence Handle14707166
GC Conant A Wagner (2003) ArticleTitleAsymmetric sequence divergence of duplicate genes Genome Res 13 2052–2058 Occurrence Handle1:CAS:528:DC%2BD3sXnslKht7o%3D Occurrence Handle12952876
P Dehal et al. (2002) ArticleTitleThe draft genome of Ciona intestinalis: insights into chordate and vertebrate origins Science 298 2157–2167 Occurrence Handle1:CAS:528:DC%2BD38XpsVSkt7o%3D Occurrence Handle12481130
M Featherstone (2003) HOX proteins and their co-factors in trancriptional regulation T Lufkin (Eds) Murine homeobox gene control of embryonic patterning and organogenesis Elsevier Amsterdam, 1––42 pp
DX C Ferrier Minguillón PWH Holland J Garcia-Fernández (2000) ArticleTitleThe amphioxus Hox cluster: deuterostome posterior flexibility and Hox 14 Evol Dev 2 284–293 Occurrence Handle11252557
C Fried SJ Prohaska PF Stadler (2003) ArticleTitleIndependent Hox-cluster duplications in Lampreys J Exp Zool (Mol Dev Evol) 299B 18–25 Occurrence Handle1:CAS:528:DC%2BD3sXoslOrtbg%3D
C Fried SJ Prohaska PF Stadler (2004) ArticleTitleExclusion ofrepetitive DNA elements from gnathostome Hox clusters J Exp Zool (Mol Dev Evol) 302B 165–173 Occurrence Handle1:CAS:528:DC%2BD2cXjvVGqsrc%3D
L Guo-Qing MVH Wilson (1996) Phylogeny of Osteoglossomorpha M Stiassny L Parenti GD Johnson (Eds) Interrelationships of fishes Academic Press San Diego CA, 163––174
H Haack P Gruss (1993) ArticleTitleThe establishment of murine Hox-1 expression domains durine patterning of limb Dev Biol 157 410–422 Occurrence Handle1:CAS:528:DyaK3sXkt1Ght7w%3D Occurrence Handle8099045
PW Holland J Garcia-Fernandez (1996) ArticleTitleHox genes and chordate evolution Dev Biol 173 382–395 Occurrence Handle1:CAS:528:DyaK28Xps1Cktg%3D%3D Occurrence Handle8605999
P W H Holland, J Garcia-Fernández, N A Williams, A Sidow (1994) Gene duplication and the origins of vertebrate developmentDevelopment Suppl 125 133
JG Inoue M Miya K Tsukamoto M Nishida (2003) ArticleTitleBasal actinopterygian realtionships: a mitogenomic perspective on the phylogeny of the “ancient fish.” Mol Phylogenet Evol 26 110–120 Occurrence Handle1:CAS:528:DC%2BD38Xps1Wht7c%3D Occurrence Handle12470943
FY Ji JD Liu MS Yi L Huang F Zhou QX Yu (2002) ArticleTitleChromosomal localization of rice filed eel Hox genes by PRINS Acta Genet Sinica 29 612–615 Occurrence Handle1:CAS:528:DC%2BD3sXksFOqug%3D%3D
C Kappen K Schugart FH Ruddle (1989) ArticleTitleTwo steps in the evolution of Antennapedia-class vertebrate homeobox genes Proc Natl Acad Sci USA 86 5459–5463 Occurrence Handle1:CAS:528:DyaL1MXkvFWmu74%3D Occurrence Handle2568634
M Kellis BW Birren ES Lander (2004) ArticleTitleProof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae Nature 428 617–624 Occurrence Handle1:CAS:528:DC%2BD2cXivVGmtLY%3D Occurrence Handle15004568
C-B Kim C Amemiya W Bailey K Kawasaki J Mezey W Miller S Minoshima N Shimizu GP Wagner F Ruddle (2000) ArticleTitleHox cluster genomicsin the horn shark. Heterodontus francisci Proc Natl Acad Sci USA 97 1655–1660 Occurrence Handle1:CAS:528:DC%2BD3cXhsFCrsbg%3D Occurrence Handle10677514
A Kobayashi RR Behringer (2003) ArticleTitleDevelopmental genetics of the female reproductive tract in mammals Nature Rev Genet 4 969–980 Occurrence Handle1:CAS:528:DC%2BD3sXptFait7g%3D
FA Kondrashov IB Rogozin YI Wolf EV Koonin (2002) ArticleTitleSelection in the evolution of gene duplications Genome Biol 3 Occurrence Handle1:CAS:528:DC%2BD38XisVaqu7g%3D
J Krieger PA Fuerst (2002) ArticleTitleEvidence for a slowed rate of molecular evolution in the order Acipenseriformes Mol Biol Evol 19 891–897 Occurrence Handle1:CAS:528:DC%2BD38Xks1Ojtbs%3D Occurrence Handle12032245
M Lynch JS Conery (2000) ArticleTitleThe evolutionary fate and consequences of duplicate genes Science 290 1151–1155 Occurrence Handle10.1126/science.290.5494.1151 Occurrence Handle1:CAS:528:DC%2BD3cXotVChsb8%3D Occurrence Handle11073452
E Malaga-Trillo A Meyer (2001) ArticleTitleGenome duplication and accelerated evolution of Hox genes and cluster architecture in teleost fishes Am Zool 41 676–686 Occurrence Handle1:CAS:528:DC%2BD3sXhsFahtr0%3D
P Martinez CT Amemiya (2002) ArticleTitleGenomics of the HOX gene cluster Comp Biochem Physiol B 133 571–580 Occurrence Handle12470820
TJS Merrit JM Quattro (2003) ArticleTitleEvolution of the vertebrate cytosolic malate dehydrogenase gene family: duplication and divergence in actinopterygian fish J Mol Evol 56 265–276 Occurrence Handle1:CAS:528:DC%2BD3sXhslWiu7Y%3D Occurrence Handle12612830
BD Metscher, K Takahashi, K Crow, C Amemiya, DF Nonaka, GP Wagner (2005) Expression of Hoxa-11 and Hoxa-13 in the pectoral fin of a basal rayfinned fish, Polyodon spathula: implications for the origin of tetrapod limbsEvol Dev (in press)
A Meyer E Malaga-Trillo (1999) ArticleTitleVertebrate genomics: more fishy tales about Hox genes Curr Biol 9 210–213
A Meyer M Schartl (1999) ArticleTitleGene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions Curr Opin Cell Biol 11 699–704 Occurrence Handle1:CAS:528:DyaK1MXotVKjsb0%3D Occurrence Handle10600714
BY Misof GP Wagner (1996) ArticleTitleEvidence for four Hox clusters in the killifish, Fundulus heteroclitus (Teleostei) Mol Phylogenet Evol 5 309–322 Occurrence Handle1:CAS:528:DyaK28XjtVCkuro%3D Occurrence Handle8728389
B Morgenstern (1999) ArticleTitleDIALIGN 2:improvement of the segment-to-segment approach to multiple sequence alignment Bioinformatics 15 21–218
SV Muse BS Gaut (1994) ArticleTitleA likelihood approach for comparing synonymous and non-synonymous nucleotide substitution rates, with applications to the chloroblast genome Mol Biol Evol 11 715–724 Occurrence Handle1:CAS:528:DyaK2cXlvFOjsL8%3D Occurrence Handle7968485
K Naruse S Fukamachi H Mitani M Kondo T Matsuoka S Kondo N Hanmura Y Morita K Hasegawa R Nishigaki A Shimada H Wada T Kusakabe N Suzuki M Kinoshita A Kanamori T Terado H Kimura M Nonaka A Shima (2000) ArticleTitleA detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution Genetics 154 1773–1784 Occurrence Handle1:CAS:528:DC%2BD3cXjtVams70%3D Occurrence Handle10747068
K Naruse M Tanaka K Mita A Shima J Postleithwait H Mitani (2004) ArticleTitleA medaka gene map: the trace of ancestral vertebrate proto-chromosomes revealed by compariative gene mapping Genome Res 14 820–828 Occurrence Handle1:CAS:528:DC%2BD2cXjvFyktbk%3D Occurrence Handle15078856
S Ohno (1970) Evolution by Gene Duplication Springer Verl New York
AM Pavell EJ Stellwag (1994) ArticleTitleSurvey of Hox-like genes in the teleost Morone saxatilis: implications for evolution of the Hox gene family Mol Mar Biol Biotech 3 149–157 Occurrence Handle1:CAS:528:DyaK2MXmtFGkug%3D%3D
YVd Peer JS Taylor I Braasch A Meyer (2001) ArticleTitleThe ghost of selection past: rates of evolution and functinal divergence of anciently duplicated genes J Mol Evol 53 436–446 Occurrence Handle11675603
TP Powers CT Amemiya (2004) ArticleTitleEvidence for a Hox 14 paralog group in vertebrates CurrBiol 14 R183–R184 Occurrence Handle1:CAS:528:DC%2BD2cXisVeksLw%3D
V Prince FB Pickett (2002) ArticleTitleSplitting pairs: the diverging fates of duplicated genes Nat Rev Genet 3 827–837 Occurrence Handle1:CAS:528:DC%2BD38Xotlersb0%3D Occurrence Handle12415313
SJ Prohaska C Fried C Flamm GP Wagner PF Stadler (2004) ArticleTitleSurveying phylogenetic footprints in large gene clusters: application to Hox cluster duplications Mol Phylogenet Evol 31 581–604 Occurrence Handle1:CAS:528:DC%2BD2cXivVKntb8%3D Occurrence Handle15062796
SJ Prohaska PF Stadler (2004) ArticleTitleThe duplication of the Hox gene clusters in teleost fishes Tlieory Biosci 123 89–110 Occurrence Handle1:CAS:528:DC%2BD2cXnsFyhtb0%3D
M Robinson-Rechavi D Huchon (2000) ArticleTitleRRTree: relative-rate tests between groups of sequences on a phylogenetic tree Bioinformatics 16 292–297 Occurrence Handle10869024
M Robinson-Rechavi V Laudet (2001) ArticleTitleEvolutionary rates of duplicated genes in fish and mammals Mol Biol Evol 18 681–683 Occurrence Handle1:CAS:528:DC%2BD3MXis1eitrs%3D Occurrence Handle11264421
FH Ruddle JL Bartels KL Bentley C Kappen MT Murta JW Pendelton (1994a) ArticleTitleEvolution of Hox genes Ann Rev Genet 28 423–442 Occurrence Handle1:CAS:528:DyaK2MXislWlsr4%3D
FH Ruddle KL Bentley MT Murtha N Risch (1994b) ArticleTitleGene loss and gain in the evolution of the vertebrates Development IssueIDSuppl 155–161
G Ruvkun O Hobert (1998) ArticleTitleThe taxonomy of developmental control in Caenorhabditis elegans Science 282 2033–2041 Occurrence Handle1:CAS:528:DyaK1cXotVOrt7s%3D Occurrence Handle9851920
S Santini JL Boore A Meyer (2003) ArticleTitleEvolutionary conservation of regulatory elements in vertebrate Hox gene clusters Genome Res 13 1111–1122 Occurrence Handle1:CAS:528:DC%2BD3sXksFehu7Y%3D Occurrence Handle12799348
G Schlosser (2002) ArticleTitleModularity and the units of evolution Theory Biosci 121 1–80
H-C Seo R Edvardsen AD Maeland M Bjordal MF Jensen A Hansen M Flaat J Weissenbach H Lehrach P Wincker R Reinhardt D Chourrout (2004) ArticleTitleHox clouster disintegration with persitent auteroposterior order of expression in Oikopleura dioica Nature 431 67–71 Occurrence Handle1:CAS:528:DC%2BD2cXntFClu7s%3D Occurrence Handle15343333
EA Snell JL Scemama EJ Stellwag (1999) ArticleTitleGenomic organization of the Hoxa-4–Hoxa10 region from Morone saxatilis: implications for Hox gene evolution among vertebrates J Exp Zool (Mol Dev Evol) 285 41–49 Occurrence Handle1:CAS:528:DyaK1MXivF2iuro%3D
PF Stadler C Fried SJ Prohaska WJ Bailey BY Misof FH Ruddle GP Wagner (2004) ArticleTitleEvidence for independent Hox gene duplications in the hagfish lineage: a PCR-based gene inventory of Eptatretus stoutii Mol Phylogenet Evol 32 686–694 Occurrence Handle1:CAS:528:DC%2BD2cXmt1Cns7o%3D Occurrence Handle15288047
HS Taylor HG Vanden PA Igarashi (1997) ArticleTitleA conserved Hox axis in the mouse and human reproductive system: late establishment and persistent expression of the Hoxa cluster genes Biol Reprod 57 1338–1345 Occurrence Handle1:CAS:528:DyaK2sXnvVWns7w%3D Occurrence Handle9408238
JS Taylor I Braasch T Frickey A Meyer Peer YVd (2003) ArticleTitleGenome duplication, a trait shared by 22,000 species of ray-finned fish Genome Res 13 382–390 Occurrence Handle1:CAS:528:DC%2BD3sXit1Wgtrc%3D Occurrence Handle12618368
K Vanderpoele WD Vos JS Taylor A Meyer Peer YVd (2004) ArticleTitleMajor events in the genome evolution of vertebrates: paranome age and size differ considerably between ray-finned fishes and land vertebrates Proc Natl Acad Sci USA 101 1638–1643 Occurrence Handle14757817
GP Wagner C Amemiya F Ruddle (2003) ArticleTitleHox cluster duplications and the opportunity for evolutionary novelties Proc Natl Acad Sci USA 100 14603–14606 Occurrence Handle1:CAS:528:DC%2BD3sXpvFaru70%3D Occurrence Handle14638945
GP Wagner C Fried SJ Prohaska PF Stadler (2004) ArticleTitleDivergence of conserved non-coding sequences: rate estimates and relative rate tests Mol Biol Evol 21 2116–2121 Occurrence Handle1:CAS:528:DC%2BD2cXptVKns7c%3D Occurrence Handle15282332
F Wright (1990) ArticleTitleThe ‘effective number of codons’ used in a gene Gene 87 23–29 Occurrence Handle10.1016/0378-1119(90)90491-9 Occurrence Handle1:CAS:528:DyaK3cXktVWmsbo%3D Occurrence Handle2110097
Z Yang (1997) ArticleTitlePAML: a program package for phylogenetic analysis by maximum likelihood CABIOS 13 555–556 Occurrence Handle1:CAS:528:DyaK2sXntlGnu7s%3D Occurrence Handle9367129
P Zhang Z Gu WH Li (2003) ArticleTitleDifferent evolutionary patterns between young duplicate genes in the human genome Genome Biol 4 R56 Occurrence Handle12952535
Acknowledgments
We thank Dr. Gavin Conant for reading an early version of this manuscript. The financial support by NSF Grant INB-0321470 to G.P.W. and the DFG Bioinformatics Initiative BIZ-6/1-2 to P.E.S. is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Axel Meyer
Rights and permissions
About this article
Cite this article
Wagner, G.P., Takahashi, K., Lynch, V. et al. Molecular Evolution of Duplicated Ray Finned Fish HoxA Clusters: Increased Synonymous Substitution Rate and Asymmetrical Co-divergence of Coding and Non-coding Sequences. J Mol Evol 60, 665–676 (2005). https://doi.org/10.1007/s00239-004-0252-z
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0252-z