
Abstract
Peptide synthesis in eukaryotes terminates when eukaryotic release factor 1 (eRF1) binds to an mRNA stop codon and occupies the ribosomal A site. Domain 1 of the eRF1 protein has been implicated in stop codon recognition in a number of experimental studies. In order to further pinpoint the residues of this protein involved in stop codon recognition, we sequenced and compared eRF1 genes from a variety of ciliated protozoan species. We then performed a series of computational analyses to evaluate the conservation, accessibility, and structural environment of each amino acid located in domain 1. With this new dataset and methodology, we were able to identify eight specific amino acid sites important for stop codon recognition and also to propose a set of cooperative paired substitutions that may underlie stop codon reassignment. Our results are more consistent with current experimental data than previously described models.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Termination of translation is the final step of protein biosynthesis. When translocation of the mRNA places a stop codon at the aminoacyl (A) site of the ribosome, polypeptide release factors (RFs) promote release of the nascent peptide chain. After more than 30 years of investigation, it is now known that two classes of RFs are required for translation termination. Class I release factors recognize the stop codon in the A site of the ribosome and promote hydrolysis of the ester bond linking the polypeptide chain with the peptidyl (P) site tRNA. Class II release factors function independently of the stop codon and serve to stimulate Class I release factor activity. Bacteria utilize two Class I RF homologs for termination: RF1 recognizes UAA and UAG codons, while RF2 recognizes UAA and UGA codons. In contrast, eukaryotes use a single, omnipotent release factor (eRF1) that is able to recognize all three stop codons. (Nakamura and Ito 1998; Nakamura et al. 2000; Song et al. 2000).
Recently, extensive mutational and biochemical studies in Escherichia coli have indicated that the tripeptide motifs of RF1 and RF2 function in a manner similar to tRNA anticodons. The first and the third amino acids of these motifs can discriminate between the second and the third purine bases of the codon, respectively (Ito et al. 2000). While studies in eubacterial systems have made significant progress, the underlying mechanism of stop codon recognition in eukaryotes still remains unclear. This mechanism has recently attracted a wide interest, and a number of experiments have been performed to determine how stop codon recognition is accomplished by eRF1. Protein–RNA crosslinking experiments have revealed an intimate contact between eRF1 and stop codons (Chavatte et al. 2001, 2002). Mutagenesis of eRF1 has also resulted in the modification of their stop codon recognition pattern (Bertram et al. 2000; Frolova et al. 2002; Seit-Nebi et al. 2002). All the evidence points to a direct recognition model for eRF1 stop codon discrimination.
An attractive hypothesis has been postulated in which Class I RFs mimic tRNA molecules as they bind to the ribosome, in terms of both their functions and their tertiary structures (Ito et al. 1996). This hypothesis is supported by a protein crystallography study of human eRF1, which revealed a three-domain structure, similar in its dimensions and shape to the “L”-like structure of a tRNA molecule (Song et al. 2000). Other studies have also unraveled the precise functions of different domains of eRF1: domain 1, which comprises about 140 N-terminal amino acids of eRF1, corresponds to the anticodon loop in tRNA and represents a stop codon decoding region (Bertram et al. 2000); domain 2, the middle domain of eRF1, corresponds to the CCA stem in tRNA and contains a perfectly conserved “GGQ” motif and functions to promote the ribosomal peptidyltransferase activity (Frolova et al. 1999; Song et al. 2000); domain 3, the C-terminal residues of the protein, contains an eRF3-binding site (Ito et al. 1998).
Several alternative models for stop codon recognition by eRF1 domain 1 have been published, based on either mutational study or comparative sequence analysis (Bertram et al. 2000; Inagaki et al. 2002; Muramatsu et al. 2001; Nakamura et al. 2000). As a result, more than 14 stop codon-binding sites have been proposed in the literature. However, none of these models is entirely consistent with recent experimental evidence. Therefore, in order to further define the stop codon-binding residues of eRF1 domain 1, we have analyzed the conservation and three-dimensional properties of all amino acids in this region of the protein. To aid in this analysis, we sequenced the eRF1 genes from six species of stichotrichous ciliates that reassign the stop codons UAA and UAG to glutamine and from the anaerobic ciliate Nyctotherus ovalis, which uses the standard code (van Hoek et al. 1998; Lozupone et al. 2001). A series of in vitro and in vivo experiments has demonstrated that the eRF1 proteins in other ciliate species have lost the ability to recognize the reassigned stop codons (Ito et al. 2002; Kervestin et al. 2001). Using this augmented dataset of eRF1 proteins, we have identified candidate residues involved in stop codon recognition.
Materials and Methods
Cloning and Sequencing of Ciliate eRF1 Genes
We designed degenerate primers to conserved regions of the eRF1 protein flanking the codon recognition domain (5′-GTNRRKY TSCCHAARAARCAYRGAARRGAA-3′ and 5′-GCYTSRYT NARWCCATTYTCKCCWCCRTA-3′) and performed PCR to amplify the intervening region of the eRF1 gene from seven ciliates species. The species we chose for this study were Paraurostyla weissei, Uroleptus sp., Urostyla grandis, Holosticha sp., Gonostomum sp., Eschaneustyla sp., and Nyctotherus ovalis. Urostyla grandis DNA was a gift from David Prescott (Department of Molecular, Cellular and Developmental Biology, University of Colorado, Boulder). N. ovalis cells were a gift from J. Hackstein (Department of Evolutionary Microbiology, University of Nijmegen, the Netherlands). The others, a gift from Wei-Jen Chang (Ecology and Evolutionary Biology, Princeton University, Princeton, NJ) and Mann-Kyoon Shin (Department of Biological Science, University of Ulsan, Ulsan, Korea), were isolated from local Princeton lakes and soils and characterized by morphology to the genus level. Paraurostyla and Uroleptus identification was confirmed by small subunit rDNA sequence comparison to related sequences in GenBank (Chang and Shin, unpublished results).
We amplified the 5′ and 3′ ends of the macronuclear chromosomes containing the eRF1 genes using gene-specific and telomere-specific primers in a variation on PCR called telomere suppression PCR, as described elsewhere (Curtis and Landweber 1999). The PCR products were visualized on a 1% agarose gel, and bands corresponding to the predicted sizes were cut from the gel and purified using Genelute Minus EtBr Spin Columns (Sigma) according to the manufacturer’s protocol. The products were cloned using the TOPO TA Cloning Kit (Invitrogen), and multiple clones were sequenced at the Syn/Seq Facility of Princeton University.
Identification of Important Sites in Domain 1
We created an alignment of the eRF1 protein sequences available in GenBank (43 sequences), plus the seven newly identified ciliate genes, using ClustalX (Version 1.81) (Thompson et al. 1994), with all settings left at default value.
We used the Evolutionary Trace Method (Lichtarge et al. 1996) to study amino acid conservation at each position in the protein alignment. First, the eRF1 protein sequences were divided into three groups based on stop codon usage: the UGA group (ciliates that use only UGA as a stop codon, including six in this study), the UAR group (Euplotes and Blepharisma, which use only UAG and UAA as stop codons [Lozupone et al. 2001]), and the standard group (in which eRF1 recognizes all three stop codons). N. ovalis avoids use of UGA codons (J. Hackstein et al., unpublished data; see below), making its eRF1 specificity ambiguous, so we considered it a separate group. A consensus sequence of “conserved” positions was assembled for each group, based on amino acids found to be identical in the subalignment, with all other sites designated “neutral.” The consensus sequences from each of the three groups were then aligned to obtain the evolutionary trace for the entire group. In the trace, a position was designated neutral if it was neutral in any of the consensus sequences. A position was designated conserved if all consensus sequences had the same invariant residue at that position or was considered “class-specific” if it varied only between groups.
The program ENVIRON (Koehl and Delarue 1994) was used to study the accessibility of each amino acid position of a representative eRF1 protein. ENVIRON is a program designed to calculate the accessibility of amino acid positions at the surface of the protein. The PDB file of human eRF1 protein (1DT9) was used as a model, and the accessible surface area of each residue was calculated. If ENVIRON determined the percentage of an amino acid’s accessible surface area to be greater than 25% of the free surface area (the free surface area of amino acid x is defined as the surface area of amino acid x in a Gly–x–Gly tripeptide), that amino acid was identified as an external residue and marked as 1. Otherwise, it was identified as an internal residue and marked as 0.
Three-dimensional (3D) cluster analysis was finally used to study the structural environment of each amino acid (Landgraf et al. 2001). Human eRF1 was used as a reference structure for the eRF1 family of proteins in this investigation. A (global) multiple sequence alignment was created containing representatives of the standard group. For each amino acid in the reference structure we identified its neighbors, defined as those amino acids whose Cα lies within a sphere of radius 10 \(\AA \) centered in the Cα of the chosen amino acid. A regional alignment containing the structural neighbors for each residue in domain 1 was extracted from the global alignment. Global and regional similarity matrices containing the pairwise sequence similarity scores for all sequences within the respective alignment were calculated. Finally, the regional conservation score for each residue in the reference structure was calculated. The score represents the difference in conservation between the structural neighbors of each residue and the protein as a whole. The higher the conservation score is, the more conserved is the cluster of structural neighbors. The threshold 1.4 (P value <0.02) was chosen, and the residues with a higher score were identified.
Results
Characteristics of Novel eRF1 Genes
We determined the complete macronuclear sequence of the Paraurostyla weissei, Uroleptus sp., Urostyla grandis, Holosticha sp., Gonostomum sp., Eschaneustyla sp., and Nyctotherus ovalis eRF1 genes (GenBank accession numbers: AY517520–AY517527). Each gene can encode a protein of approximately 450 amino acids, typical for eRF1 proteins, which range in size from 410 to 460 amino acids. The identity of these ciliate proteins to the human eRF1 protein ranges from 57 to 60%, while the similarity to the human eRF1 ranges from 73 to 77% (Table 1). The P. weissei, Uroleptus sp., Urostyla grandis, Gonostomum sp., and Nyctotherus ovalis sequences appear to contain introns matching the stichotrich intron consensus sequence (Prescott et al. 2002) at conserved locations at the 5′ end of the gene. These introns were removed to facilitate translation. In-frame UAR codons present in all these eRF1 gene sequences except Nyctotherus ovalis further confirmed their predicted stop codon usage profile.
Stop Codon-Binding Sites in eRF1 Domain 1
We evaluated each amino acid position of the eRF1 protein for three features: conservation, accessibility to other macromolecules, and structural environment.
Conservation
Because of the evolutionary pressure to maintain their functional integrity, homologous proteins undergo fewer substitutions at binding site residues than at other, less functionally important amino acid sites (Lichtarge et al. 1996). Substitutions at these positions are likely to cause functional divergence. We used the Evolutionary Trace Method (Lichtarge et al. 1996) to determine the conservation profile of the amino acid positions. We found 29 amino acid sites conserved among all species and 4 amino acid sites showing class-specific profiles. Figure 1 shows the position of these residues in an alignment of domain 1 for the 51 sequences used in this study.

Results of Evolutionary Trace Method and ENVIRON program. Available eRF1 protein sequences were collected from public databases, aligned, and then classified according to the stop codon usage of the organism. Each type of amino acid is shown in a specific color. Amino acids with similar chemical properties are shown in similar colors. Amino acid positions are numbered according to the human eRF1 sequence. Above the alignment, surface (accessible) positions are marked with a 1; internal (inaccessible) positions are marked with a 0. For evolutionary trace, neutral positions are left blank; conserved positions are shown by one-letter amino acid abbreviations, and class-specific positions are identified with an X. Putative stop codon-binding residues inferred in this study are marked by a vertical line and class-specific residues are in boldface. Conserved and class-specific residues are highlighted. Proteins are named using a three-letter species identifier: Tth, Tetrahymena thermophila AF298833; Pte, Paramecium tetraurelia AAK66860; Par, Paraurostyla weissei AY517520 (this study); Gon, Gonostomum sp. AY517524 (this study); Hol, Holosticha sp. AY517523 (this study); Esc, Eschaneustyla sp. AY517525 (this study); Url, Uroleptus sp. AY517521 (this study); Uro, Urostyla grandis AY517522 (this study); Otr, Oxytricha trifallax Q9BMX3; Ono, Oxytricha nova (AF188150; this sequence is a single randomly sampled clone that contains some errors); Sle, Stylonychia lemnae Q9BMMO; Smy, Stylonychia mytilus Q9BMM1; Sal, Stichotrichida sp. Alaska AAN62564; Tpu, Tetmemena pustulata AAN62568; EaeA, Euplotes aediculatus A AF298831; EaeB, Euplotes aediculatus B AF298832; EocA, Euplotes octocarinatus A CAC14170; EocB, Euplotes octocarinatus B AAG25924; Bja, Blepharisma japonicum CAC16186; Bam, Blepharisma americanum Q9BMM3; NovA, Nyctotherus ovalis A AY517526 (this study); NovB, Nyctotherus ovalis B AY517527 (this study); Pfa, Plasmodium falciparum AAC71899; Pyo, Plasmodium yoelii; Gth, Guillardia theta AF165818; Gin, Giardia intestinalis AF198017; Lma, Leishmania major CAB77686; Tbr, Trypanosoma brucei AF278718; Ani, Aspergillus nidulans AF451327; Ncr, Neurospora crassa AAL17659; Pan, Podospora anserine AAC08410; Pca, Pneumocystis carinii BAB61041; Cal, Candida albicans; Sce, Saccharomyces cerevisiae CAA51935; Spo, Schizosaccharomyces pombe P79063; Dme, Drosophila melanogaster AAF51574; Xla, Xenopus laevis P35615; Has, Homo sapiens P46055; Mau, Mesocricetus auratus CAA57282; Mmu, Mus musculus_NM 144866; Pmi, Polyandrocarpa misakiensis Q9GR88; Cel, Caenorhabditis elegans T31907; Ecu, Encephalitozoon cuniculi CAD26553; Ddi, Dictyostelium discoideum AF298834; Tva, Trichomonas vaginalis AAL17661; Cre, Chlamydomonas reinhardtii AAL17660; Ath1, Arabidopsis thaliana 1 AAA91169; Ath2, Arabidopsis thaliana 2 AC012187; Ath3, Arabidopsis thaliana 3 CAA49172; Osa, Oryza sativa BAB89728.
Accessibility
Because all eRF1 homologs show high sequence similarity, the surface accessibility of each amino acid in a homolog of eRF1 can be predicted by determining the accessibility of its homologous site in a reference structure. Based on the structure determined for the human eRF1 domain 1, residues in this domain can be classified as internal residues (<25% accessible free surface area) or external residues (>25% accessible free surface area). While internal residues can be important to the structural stability of the protein, they are unlikely to interact physically with other macromolecules. Figure 1 shows the position of the 67 accessible amino acid sites in the domain 1 alignment.
Structural Environment
Protein interaction sites often comprise more than one residue. These residues need not lie adjacent to one another in the primary sequence of the protein, since distant positions are often juxtaposed once the protein is properly folded. In addition, specific properties of a binding site residue, such as charge and conformation, can be strongly influenced by neighboring residues. As a result, binding sites tend to be located in relatively conserved structural environments in order to preserve their biological functions. It has been shown that the detection of functional residues can be greatly enhanced when their structural neighbors are considered (Landgraf et al. 2001). We found that 20 amino acid sites lie in a highly conserved structural environment (Fig. 2).

Three-dimensional cluster analysis of external residues. The relative conservation of the structural environment surrounding each amino acid position relative to the human eRF1 protein. The higher the conservation score, the more conserved is the cluster of structural neighbors. The conservation score of all the internal positions is set to 0.
Discussion
Previous comparative eRF1 sequence analyses were based on a limited number of eRF1 sequences in species with variant genetic codes, making the identification of stop codon-binding sites very difficult. Here we increased the number of biologically meaningful eRF1 sequences and used the Evolutionary Trace Method (Lichtarge et al. 1996) to evaluate the conservation of each amino acid in domain 1 in three different stop codon usage groups. Thus some sites that show great diversity could be ruled out.
Our study also takes full advantage of the structural information available for the eRF1 protein by considering the accessibility and local structural environment of each amino acid. Based on the 3D structure of domain 1 in eRF1, we classified the residues into external and internal sites. Moreover, we also performed 3D cluster analysis (Landgraf et al. 2001) to evaluate the local structural environment of each residue in domain 1. Because the difference between stop codons and a near-cognate codon is subtle, we reasoned that it is more likely that residues in a relatively conserved environment provide such an accurate interaction.
Although several authors propose conformational changes of eRF1 upon binding to the ribosomal A site (Chavatte et al. 2002; Inagaki et al. 2002), such changes would not have a large impact on our results according to a recent crosslinking study (Chavatte et al. 2003a). This study suggested a two-step model for eRF1 binding to the A site: a codon-independent step is followed by a stop codon-dependent isomerization step. Direct recognition of the stop codons by domain 1 of eRF1 triggers a rearrangement of bound eRF1 from an open to a closed conformation, allowing the universally conserved GGQ motif to approach the peptidyl transferase center of the ribosome. This eRF1 rearrangement is mainly an interdomain conformational change, which is likely to be achieved by manipulating a flexible peptide hinge between domain 1 and domain 2. Therefore, although full recognition of the stop codons may lead to some further minor changes, the conformation of domain 1 in the free eRF1 crystal should still largely represent the initial state of the decoding region approaching the stop codons. Thus the availability of the domain 1 3D structure provides a source of significant information which can improve the sensitivity of binding site prediction.
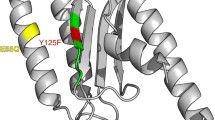
As a result, our three-pronged computational approach identified eight residues in domain 1 that are conserved, accessible at the surface of the molecule, and reside in a conserved structural environment. The fact that all these eight sites were simultaneously identified by three independent methods strengthens our confidence in the biological interpretation of our results. We consider these sites strong candidates to be responsible for the physical interaction of eRF1 with the mRNA stop codon mediating stop codon recognition. Five of these sites (31, 32, 62, 63, and 127) are absolutely conserved across all the species analyzed, while positions 57, 70, and 126 are conserved only among species employing the same set of stop codons (Fig. 3).

Important sites in eRF1 domain 1. Crystal structure of human eRF1 marked with putative stop codon-binding sites identified in this study. A The whole human eRF1 crystal structure: conserved positions are shown in orange, class-specific positions are shown in blue, and the GGQ motif is shown in red. B Human eRF1 domain 1 crystal structure: conserved positions are shown in orange, and class-specific positions are shown in blue.
These eight residues lie in a relatively small area of eRF1 domain 1, and the distance between the farthest apart of these residues (63 and 126) is about 20 \(\AA \). The distribution of these sites may be a little more scattered than expected for eRF1 to interact with the stop codon trinucleotide, whose bases are well stacked as in a helix. However, the conformational flexibility of single-stranded RNA plus the large size of an individual nucleotide (∼10 \(\AA \)) makes it physically conceivable that the proposed residues could interact together with a stop codon. More importantly, it is very likely that a local conformational shift in domain 1 during binding could bring the proposed sites even closer. Conformational shifts may play an important role in increasing the specificity and fidelity of stop codon recognition.
Most of the putative stop codon-binding amino acids identified in our study receive considerable support from experimental evidence. Residues 62 and 63 are located in the highly conserved NIKS tetrapetide which mutagenesis studies suggested is functionally essential (Frolova et al. 2002). More recently, it has been shown that the invariant undine (U), found at the first position of each stop codon, can be photocrosslinked to position K63 (and possibly to its close neighbors as well) of eRF1 when both are situated in the ribosome (Chavatte et al. 2002). This strongly suggests that position 63 is directly involved in stop codon recognition. Residue 127, located in the Y–C–F minidomain, is also believed to play a critical role in stop codon recognition, because mutations at this position resulted in peptide release activity changes (Seit-Nebi et al. 2002). Finally, mutations at positions 126 and 71 (the immediate neighbor of position 70, identified in our study) impair stop codon recognition (Bertram et al. 2000).
When we compare our results with previously described models (“anticodon mimicry model” [Muramatsu et al. 2001; Nakamura et al. 2000] and “cavity-binding model” [Bertram et al. 2000; Inagaki et al. 2002]), we observe some overlap in the proposed residues. However, our results appear to be more compatible with current experimental results, especially residues 62 and 63, which were experimentally determined to be important (Chavatte et al. 2002), and are identified in our sequence-based analysis but were not included in any previous model.
Our results suggest three class-specific positions. Substitutions at these positions may be responsible for the inability of some ciliate species to terminate translation at UGA and, for others, including the six stichotrichs in this study, UAR. As demonstrated in Table 2, the substitution pair G57S and L126F is associated with UAR reassignment, while S70A and L126I are associated with UGA reassignment. In the UGA stop codon group, Tetrahymena/Paramecium is phylogenetically distinct from the Stylonychia group (Hewitt et al. 2003; Lozupone et al. 2001). Therefore, our results point to patterns in evolvability of the translation apparatus (Knight et al. 2001) such that key substitutions in eRF1 arose independently in different lineages, permitting reassignment of UAR or UGA from stop to sense. The presence of these striking convergent substitutions, against the background of accumulated substitutions, was also essential for the success of our computational methods.
It should be emphasized that any substitution in the proposed pairs is necessary but not sufficient to change eRF1-decoding ability. The idea that these paired substitutions lead to the alternation of stop codon specificity may resolve the controversy over position 126 in current experimental data. Position 126 shows convergent substitutions Leu to Phe in eRF1 from ciliates with the UGA = stop variant code (the O. nova sequence may appear to be an exception with L126D [Fig. 1], but this sequence is derived from one randomly sampled clone in a preliminary genome survey [Prescott et al. 2002] and has not been verified). Moreover, the release activity of these mutants has been shown to create “unipotent” eRF1 proteins that favor recognition of two of the stop codons, implying that this residue is critical for discrimination of the three stop codons (Bertram et al. 2000). On the other hand, the crosslink patterns of the mutant protein with L126F substitution were the same as that of the wild type (Chavatte et al. 2003b).
Experimental data also have indicated that a single mutation usually only suppresses the usage of a given stop codon and cannot totally abolish the usage of the stop codon. Thus, each substitution of the pair may work either as a positive determinant to favor binding one or two stop codons or as a negative determinant to exclude binding the other stop codon(s). At present we know only the overall effect of the paired changes on the specificity of eRF1.
Additionally, we sequenced two eRF1 genes of the anaerobic ciliate Nyctotherus ovalis (van Hoek et al. 1998). A preliminary analysis of about 1500 single reads from N. ovalis macronuclear molecules provided no examples for the presence of UGA either in-frame or as a stop codon, while it is present in introns (Johannes Hackstein et al., unpublished data, supported by EU contract QLK3-2002-02151 “CIMES”). The implication is that only UAA and UAG are used as stop codons and the usage of UGA is suppressed in this species. Thus N. ovalis may represent an intermediate between ciliates that use the standard genetic code and those that use a variant code. Although its specificity cannot be determined at this moment, it is worth noting that among three class-specific positions, both N. ovalis eRF1 protein sequences share substitution of an aromatic residue at position 126 with the UGA group, specifically sharing L126Y with Eschaneustyla in this group (Fig. 1 and Table 2). At positions 57 and 70, N. ovalis shares the same amino acids as the standard code group, consistent with its proposed position as an evolutionary intermediate. Therefore, it will be very meaningful to determine this species’ eRF1 specificity experimentally. We conjecture that UGA usage will be absent or nearly absent in this species.
Given the success of our method in detecting amino acids previously implicated in stop codon binding, we believe we may have discovered two additional conserved positions that function in this process (positions 31 and 32). However, we cannot distinguish whether these sites interact with the ribosome in addition or instead, since we cannot predict their exact roles based solely on their position or identities. Mutational studies of these positions should be undertaken to further understand the physical interactions that take place between eRF1 and the nucleotides of the stop codon.
In summary, our conclusions that five conserved amino acid sites (31, 32, 62, 63, and 127) and three class-specific sites (57, 70, and 126) in eRF1 domain 1 underlie stop codon recognition are more consistent with the available experimental evidence than previously proposed models of stop codon recognition. Additionally, we propose two new sites (31 and 32) that have not been implicated before by either comparative or experimental research, and these sites would be good candidates for further experimental confirmation. We also identified residues likely responsible for modification of eRF1 stop codon specificity, and we propose a cooperative mechanism involving pairs of substitutions. A clear picture of stop codon decoding by peptidyl release factors is still unresolved. A crystal structure of the ribosome bound to eRF1 will be necessary to understand stop codon recognition at the atomic level.
References
G Bertram HA Bell DW Ritchie G Fullerton I Stansfield (2000) ArticleTitleTerminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition RNA 6 1236–1247 Occurrence Handle1:CAS:528:DC%2BD3cXms1Cis7c%3D Occurrence Handle10999601
L Chavatte L Frolova L Kisselev A Favre (2001) ArticleTitleThe polypeptide chain release factor eRF1 specifically contacts the s(4)UGA stop codon located in the A site of eukaryotic ribosomes Eur J Biochem 268 2896–2904 Occurrence Handle1:CAS:528:DC%2BD3MXktVSkt7k%3D Occurrence Handle11358506
L Chavatte A Seit-Nebi V Dubovaya A Favre (2002) ArticleTitleThe invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome EMBO J 21 5302–5311 Occurrence Handle1:CAS:528:DC%2BD38Xns1Cms70%3D Occurrence Handle12356746
L Chavatte L Frolova P Laugaa L Kisselev A Favre (2003a) ArticleTitleStop codons and UGG promote efficient binding of the polypeptide release factor eRF1 to the ribosomal A site J Mol Biol 331 745–758 Occurrence Handle1:CAS:528:DC%2BD3sXlvFGgu7w%3D
L Chavatte S Kervestin A Favre O Jean-Jean (2003b) ArticleTitleStop codon selection in eukaryotic translation termination: comparison of the discriminating potential between human and ciliate eRF1s EMBO J 22 1644–1653 Occurrence Handle1:CAS:528:DC%2BD3sXisFeqsbg%3D
EA Curtis LF Landweber (1999) ArticleTitleEvolution of gene scrambling in ciliate micronuclear genes Ann NY Acad Sci 870 349–350 Occurrence Handle1:CAS:528:DyaK1MXkslGmsrk%3D Occurrence Handle10415495
LY Frolova RY Tsivkovskii GF Sivolobova NY Oparina OI Serpinsky VM Blinov SI Tatkov LL Kisselev (1999) ArticleTitleMutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis RNA 5 1014–1020 Occurrence Handle1:CAS:528:DyaK1MXltFCmsrs%3D Occurrence Handle10445876
L Frolova A Seit-Nebi L Kisselev (2002) ArticleTitleHighly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1 RNA 8 129–136 Occurrence Handle1:CAS:528:DC%2BD38Xis1WltL0%3D Occurrence Handle11911360
EA Hewitt KM Muller J Cannone DJ Hogan R Gutell DM Prescott (2003) ArticleTitlePhylogenetic relationships among 28 spirotrichous ciliates documented by rDNA Mol Phylogenet Evol 29 258–267 Occurrence Handle1:CAS:528:DC%2BD3sXnt1amtr8%3D Occurrence Handle13678681
Y Inagaki C Blouin WF Doolittle AJ Roger (2002) ArticleTitleConvergence and constraint in eukaryotic release factor 1 (eRF1) domain 1: the evolution of stop codon specificity Nucleic Acids Res 30 532–544 Occurrence Handle1:CAS:528:DC%2BD38XhtFCgt7k%3D Occurrence Handle11788716
K Ito K Ebihara M Uno Y Nakamura (1996) ArticleTitleConserved motifs in prokaryotic and eukaryotic polypeptide release factors: tRNA-protein mimicry hypothesis Proc Natl Acad Sci USA 93 5443–5448 Occurrence Handle1:CAS:528:DyaK28Xjt1Ojs7c%3D Occurrence Handle8643594
K Ito K Ebihara Y Nakamura (1998) ArticleTitleThe stretch of C-terminal acidic amino acids of translational release factor eRF1 is a primary binding site for eRF3 of fission yeast RNA 4 958–972 Occurrence Handle1:CAS:528:DyaK1cXltFykt78%3D Occurrence Handle9701287
K Ito M Uno Y Nakamura (2000) ArticleTitleA tripeptide ‘anticodon’ deciphers stop codons in messenger RNA Nature 403 680–684 Occurrence Handle1:CAS:528:DC%2BD3cXht1yjurs%3D Occurrence Handle10688208
K Ito L Frolova A Seit-Nebi A Karamyshev L Kisselev Y Nakamura (2002) ArticleTitleOmnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1 Proc Natl Acad Sci USA 99 8494–8499 Occurrence Handle1:CAS:528:DC%2BD38XltF2iu7k%3D Occurrence Handle12084909
S Kervestin L Frolova L Kisselev O Jean-Jean (2001) ArticleTitleStop codon recognition in ciliates: Euplotes release factor does not respond to reassigned UGA codon EMBO Rep 2 680–684 Occurrence Handle1:CAS:528:DC%2BD3MXmt1Olsbs%3D Occurrence Handle11463747
RD Knight SJ Freeland LF Landweber (2001) ArticleTitleRewiring the keyboard: Evolvability of the genetic code Nat Rev Genet 2 49–58 Occurrence Handle1:CAS:528:DC%2BD3MXisVGjs7g%3D Occurrence Handle11253070
P Koehl M Delarue (1994) ArticleTitlePolar and nonpolar atomic environments in the protein core: implications for folding and binding Proteins 20 264–278 Occurrence Handle1:CAS:528:DyaK2MXisVGju7k%3D Occurrence Handle7892175
R Landgraf I Xenarios D Eisenberg (2001) ArticleTitleThree-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins J Mol Biol 307 1487–1502 Occurrence Handle1:CAS:528:DC%2BD3MXisVKntrg%3D Occurrence Handle11292355
O Lichtarge HR Bourne FE Cohen (1996) ArticleTitleAn evolutionary trace method defines binding surfaces common to protein families J Mol Biol 257 342–358 Occurrence Handle1:CAS:528:DyaK28XhvVOqsLw%3D Occurrence Handle8609628
CA Lozupone RD Knight LF Landweber (2001) ArticleTitleThe molecular basis of nuclear genetic code change in ciliates Curr Biol 11 65–74 Occurrence Handle1:CAS:528:DC%2BD3MXhtVSgtbo%3D Occurrence Handle11231122
T Muramatsu K Heckmann C Kitanaka Y Kuchino (2001) ArticleTitleMolecular mechanism of stop codon recognition by eRF1: a wobble hypothesis for peptide anticodons FEBS Lett 488 105–109 Occurrence Handle1:CAS:528:DC%2BD3MXhsVSrs7k%3D Occurrence Handle11163755
Y Nakamura K Ito (1998) ArticleTitleHow protein reads the stop codon and terminates translation Genes Cells 3 265–278 Occurrence Handle1:CAS:528:DyaK1cXltFOqtbs%3D Occurrence Handle9685178
Y Nakamura K Ito M Ehrenberg (2000) ArticleTitleMimicry grasps reality in translation termination Cell 101 349–352 Occurrence Handle1:CAS:528:DC%2BD3cXjsFekurg%3D Occurrence Handle10830162
DM Prescott JD Prescott RM Prescott (2002) ArticleTitleCoding properties of macronuclear DNA molecules in Sterkiella nova (Oxytricha nova) Protist 153 71–77 Occurrence Handle1:CAS:528:DC%2BD38XktlSjs7k%3D Occurrence Handle12022278
A Seit-Nebi L Frolova L Kisselev (2002) ArticleTitleConversion of omnipotent translation termination factor eRF1 into ciliate-like UGA-only unipotent eRF1 EMBO Rep 3 881–886 Occurrence Handle1:CAS:528:DC%2BD38Xnt1GntLs%3D Occurrence Handle12189178
H Song P Mugnier AK Das HM Webb DR Evans MF Tuite BA Hemmings D Barford (2000) ArticleTitleThe crystal structure of human eukaryotic release factor eRF1—Mechanism of stop codon recognition and peptidyl-tRNA hydrolysis Cell 100 311–321 Occurrence Handle1:CAS:528:DC%2BD3cXhtFKjtLc%3D Occurrence Handle10676813
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 22 4673–4680 Occurrence Handle1:CAS:528:DyaK2MXitlSgu74%3D Occurrence Handle7984417
AH van Hoek TA van Alen VS Sprakel JH Hackstein GD Vogels (1998) ArticleTitleEvolution of anaerobic ciliates from the gastrointestinal tract: phylogenetic analysis of the ribosomal repeat from Nyctotherus ovalis and its relatives Mol Biol Evol 15 1195–1206 Occurrence Handle1:CAS:528:DyaK1cXls1KisL8%3D Occurrence Handle9729884
Acknowledgments
The authors would like to thank Drs. Wei-Jen Chang and Mona Singh for helpful discussions, Nicholas Stover for critical manuscript reading, and David Prescott, Johannes Hackstein, Mann-Kyoon Shin, and Wei-Jen Chang for gifts of cells or DNA. We thank two anonymous reviewers for their valuable suggestions. This work was supported by NIGMS Grant GM59708 and NSF Award 9875184 to L.F.L.
Author information
Authors and Affiliations
Corresponding author
Additional information
Han Liang, Jonathan Y. Wong,Contributed equally to this paper
ReviewingEditor: Dr. Niles Lehman
Rights and permissions
About this article
Cite this article
Liang, H., Wong, J.Y., Bao, Q. et al. Decoding the Decoding Region: Analysis of Eukaryotic Release Factor (eRF1) Stop Codon-Binding Residues. J Mol Evol 60, 337–344 (2005). https://doi.org/10.1007/s00239-004-0211-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0211-8