
Abstract
One characteristic of sex chromosomes is the accumulation of a set of different types of repetitive DNA sequences in the Y chromosomes. However, little is known about how this occurs or about how the absence of recombination affects the subsequent evolutionary fate of the repetitive sequences in the Y chromosome. Here we compare the evolutionary pathways leading to the appearance of three different families of satellite-DNA sequences within the genomes of Rumex acetosa and R. papillaris, two dioecious plant species with a complex XX/XY1Y2 sex-chromosome system. We have found that two of these families, one autosomic (the RAE730 family) and one Y-linked (the RAYSI family), arose independently from the ancestral duplication of the same 120-bp repeat unit. Conversely, a comparative analysis of the three satellite-DNA families reveals no evolutionary relationships between these two and the third, RAE180, also located in the Y chromosomes. However, we have demonstrated that, regardless of the mechanisms that gave rise to these families, satellite-DNA sequences have different evolutionary fates according to their location in different types of chromosomes. Specifically, those in the Y chromosomes have evolved at half the rate of those in the autosomes, our results supporting the hypothesis that satellite DNAs in nonrecombining Y chromosomes undergo lower rates of sequence evolution and homogenization than do satellite DNAs in autosomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sex chromosomes, while having evolved independently in several different groups of organisms, share common evolutionary features (Charlesworth 1996). Thus, the gradual suppression of recombination between the sex chromosomes is thought to lead to their progressive divergence and to the erosion of the Y chromosome (Filatov et al. 2000). The final outcome of this process should be the loss of function of many genes within the Y chromosome (Charlesworth 2002) and the degeneration of the Y chromosome, principally due to the accumulation of a set of diverse repetitive sequences such as mobile elements and satellite DNAs (Steinemann and Steinemann 1997; Bachtrog 2003a,b; Skaletsky et al. 2003). In fact, models of evolutionary dynamics for satellite DNA predict its accumulation in chromosomal regions where recombination rates are low (Charlesworth et al. 1994) and such accumulation has been documented in the nonrecombining Y chromosomes (Charlesworth 1996; Jobling and Tyler-Smith 2003; Skaletsky et al. 2003). However, little is known about how this occurs or about how the absence of recombination affects the subsequent evolutionary fate of the repetitive sequences in the Y chromosome.
Satellite DNAs are tandemly arrayed, highly repetitive DNA sequences of the eukaryotic genomes located in the constitutive heterochromatin (Ugarkovic and Plohl 2002). The repeats comprising a satellite-DNA family do not evolve independently of one another but rather follow concerted evolution (Dover 1982, 1986). That is, arrays of nonallelic homologous sequences, homogenized by transfer mechanisms such as unequal crossing-over and gene conversion, evolve as a unit (Dover 1982; Ugarkovic and Plohl 2002). Factors affecting concerted evolution include rates of transfer between homologous and nonhomologous chromosomes, arrangements of repeats, array sizes, and population structure. Bias in any of these factors can alter the rates of concerted evolution. Thus, for instance, these rates will be reduced by mechanisms impeding chromosomal exchanges (i.e., recombination). Therefore, reduced rates of concerted evolution would be expected in satellite-DNA sequences in the nonrecombining Y chromosomes.
In Rumex acetosa, a dioecious plant species with a complex sex chromosome system, females have a karyotype composed of 14 chromosomes (2n = 12 + XX), while males have 15 chromosomes (2n = 12 + XY1Y2). During meiosis, the two Y chromosomes pair only with the ends of each X arm. All the data indicate that the Ys and the X chromosomes are highly differentiated and that the Y chromosomes are degenerated, as they are heterochromatic and rich in satellite-DNA sequences (Ruiz Rejón et al. 1994). To date, three satellite-DNA families have been described within the genome of R. acetosa, two of which are present in the Y chromosomes, the RAYSI family (Shibata et al. 1999) and the RAE180 family (Shibata et al. 2000a). The RAE730 satellite-DNA family is found in heterochromatic segments of some autosome pairs (Shibata et al. 2000b). R. papillaris is a dioecious species of the same section as R. acetosa (Acetosa) within the subgenus Acetosa (Rechinger 1964), which also has a complex XX/XY1Y2 sex-chromosome system (Löve 1967). In this study, we use these species to investigate the evolutionary fate of satellite-DNA sequences according to their location in different types of chromosomes. Specifically, this work not only demonstrates the existence of common mechanisms for satellite-DNA formation and accumulation in the heterochromatic segments of autosomes and in the sex chromosomes, but also upholds the hypothesis that satellite DNAs in nonrecombining Y chromosomes undergo lower evolutionary rates than do satellite DNAs in autosomes.
Materials and Methods
Rumex acetosa was collected from Sierra Nevada (Granada), while R. papillaris was collected from Sierra de Baza (Granada). We analyzed up to six individuals from each of these two populations. DNA from leaves was extracted by using the Plant DNAzol kit (Invitrogen), following the manufacturer’s recommendations.
In both species, we studied three satellite-DNA families previously found in R. acetosa (Shibata et al. 1999, 2000a, b). The location of the three satellite-DNA families in R. papillaris was checked by in situ hybridization following the procedure described in De la Herrán et al. (2001), proving to be similar to that in R. acetosa. For the isolation of nonallelic repeats of each of the three satellite-DNA families, we followed the PCR approach using primers designated for each family. As satellite-DNA sequences are tandemly arrayed, we expected that each PCR primer pair led to the amplification of a combination of PCR products consisting of one to several adjacent repeat units or monomers. The RAE730 satellite-DNA family is the main constituent of the polymorphic heterochromatic supernumerary segments of English and Japanese populations (Shibata et al. 2000b). The supernumerary segments are fixed in homozygosis at the sixth chromosome pair in Spanish populations of Sierra Nevada and Sierra de Baza (Granada) (Ruiz Rejón et al. 1994). The primers R730-A, 5′-CTCGGACCAATT ATCTCAT-3′, and R730-B, 5′-CATTATTTGGGAGCCGAT-3′, were used to amplify the RAE730 sequences in R. acetosa and R. papillaris. The RAE180 family is one of the two major types of sequences found within the heterochromatin of the Y chromosomes (Shibata et al. 2000a). Moreover, two minute additional loci exist in euchromatic regions of the autosome pairs 1 and 4 (Shibata et al. 2000a). We obtained RAE180 sequences from males by PCR amplification with the primers R180-A, 5′-TCATCGAACTTCATTCAT-3′, and R180-B, 5′-TATAGTAATATCTCGATC-3′. The RAYSI family is exclusive of the Y chromosomes (Shibata et al. 1999). The primers RAYSI-A, 5′-ATGTAAGCATTTGGT CCTAA-3′, and RAYSI-D, 5′-TCGAGTACTACACGATTGT-3′, were designed for the amplification of RAYSI sequences. The primers RAYSI-J, 5′-GAGAGTCAATAGAGTGGAAG-3′, and RAYSI-S, 5′-ACGTAGTCTTTTAGAGGATC-3′, were each used in combination with the primer RAYSI-D to obtain sequences of each of two RAYSI subfamilies found during this study.
PCR amplifications were made in 50-μl reactions containing 10 ng of purified DNA, a 2 mM concentration of dNTPs, a 2 mM concentration of each primer, and 1.25 units of Taq polymerase in 10 mM Tris–HCl at pH 8.3, 5 mM KCl, 2 mM MgCl reaction buffer. Thermal cycles consisted of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C.
The PCR products were ligated into the plasmid pGEM-T Easy (Promega) and transformed into Escherichia coli JM109 competent cells (Promega) following the manufacturer’s instructions. Clones of each marker were sequenced by the dideoxy sequencing method using an automatic ABI-Prism 377 sequencer (Applied Biosystems). When possible, we sequenced clones containing several adjacent monomer units. The EMBL accession numbers for the sequences are AJ580328 to AJ580343, AJ580382 to AJ580398, AJ580457 to AJ580463, AJ580468 to AJ580485, AJ580494 to AJ580496, AJ634478 to AJ634526, AJ634533 to AJ63456, and AJ639709 to AJ639741.
Southern-blot hybridization analyses were conducted following the method previously reported (Garrido-Ramos et al. 1999).
For sequence analysis, multiple alignments were performed using ClustalX (Thompson et al. 1997) followed by manual adjustments. We computed basic sequence statistics with the program DnaSP version 3 (Rozas and Rozas 1999). Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 2.1 (Kumar et al. 2001). Distances were calculated according to the Jukes–Cantor method, and trees constructed by the neighbor-joining method (Saito and Nei 1987). We have followed the method proposed by Strachan et al. (1985) to analyze the pattern of variation at each nucleotide site between a pair of species in each of the three markers analyzed. This method compares the sequences obtained from two species at each site and classifies each one in any of six different transitional stages (classes 1 to 6) toward the process of full homogenization and fixation (for a detailed explanation of this method, see also Pons et al. 2002). The class 1 site represents complete homogeneity across all clones sampled from a pair of species, whereas classes 2, 3, and 4 represent intermediate stages in which one of the species shows polymorphism. The frequency of the new nucleotide variant at the site considered is low in stage 2 and intermediate in stage 3, while class 4 is composed of sites in which a mutation has replaced the progenitor base in most members of the repetitive family in the other species (almost fully homogenized site). Class 5 represents diagnostic sites in which a new variant is fully homogenized and fixed in all the members of one of the species while the other species retains the progenitor nucleotide. A class 6 site represents an additional step over stage 5 (new variants appear in some of the members of the repetitive family at a site fully divergent between the two species).
Results
Isolation, Characterization, and Origin ofSatellite-DNA Families in Rumex
We studied three satellite-DNA families in R. acetosa and R. papillaris: the Y-linked RAE180 and RAYSI families and the autosomal RAE730 family. PCR using RAE180-A and RAE180-B primers resulted in the amplification of a combination of PCR products consisting of one to four adjacent RAE180 monomers (monomers varying between 181 and 186 bp in length). However, due to the much greater length of RAYSI (922–932 bp) and RAE730 (727-731 bp) repeats, PCR-amplification experiments with primers for these two satellite DNAs gave only monomers as the PCR product. After the cloning of the amplified products, for each satellite-DNA family, we sequenced a sample of between 4 and 7 monomers from each of six individuals per species—hence, we analyzed a total of 68 RAYSI sequences (42 from R. acetosa and 26 from R. papillaris), 73 RAE180 sequences (42 from R. acetosa and 31 from R. papillaris), and 47 RAE730 sequences (21 monomeric units from R. acetosa and 26 from R. papillaris).
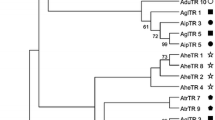
In R. acetosa, the primers RAYSI-A and RAYSI-D amplified two groups of RAYSI sequence variants. Multiple-alignment comparisons between monomeric sequences revealed the presence of 90 diagnostic sites that differentiate the two groups of sequences. Each of these 90 sites represents a particular mutation shared by all the sequences of one group, while at the same sites all the sequences of the other group had a different nucleotide. Thus, two paralogous RAYSI subfamilies can be defined in R. acetosa. We called these two RAYSI subfamilies RAYSI-S and RAYSI-J. Two new primers were then designated for the specific amplification of monomers of each RAYSI subfamily. A total of 23 RAYSI-J monomers and 19 RAYSI-S monomers were sequenced. A phylogenetic analysis of the 42 sequences reveals a neighbor-joining (NJ) tree with two clades (supported by 100% of the bootstrap replicates), one composed of RAYSI-J sequences and the other composed of RAYSI-S sequences (Fig. 1). RAYSI-S sequences were not found in R. papillaris either using the RAYSI-A/RAYSI-D combination of primers or using the subfamily-specific primers. We sequenced a total of 26 RAYSI sequences from R. papillaris, all belonging to the RAYSI-J subfamily (Fig. 1). Furthermore, Southern-blot hybridization revealed no RAYSI-S sequences, but only RAYSI-J sequences, in this species, while both subfamilies were found by this approach in R. acetosa. Therefore, for comparisons of RAYSI sequences between R. acetosa and R. papillaris (as explained below), we used only orthologous sequences from the RAYSI-J subfamily present in both species.

Neighbor-joining tree of the relationships among RAYSI sequences of R. acetosa and R. papillaris. Numbers at each node indicate the percentage of trees representing the particular node out of 1000 bootstrap replicates. Bootstrap values under 50 are not indicated. In this tree, the labels of the sequences correspond to the first three letters of the species names from which they were isolated and a number representing the repeat analyzed: ACE, R. acetosa; PAP, R. papillaris.
No subfamilies were detected in both RAE180 and RAE730 satellite DNAs. The NJ trees inferred for sequences isolated from each of these two satellite DNAs showed no indication of subfamilies either in R. acetosa or R. papillaris.
Shibata et al. (2000b) have found that the RAE730 satellite DNA originated from different cycles of amplification and sequence divergence of a basic sequence monomer of about 120 bp. We confirmed this for RAE730 sequences and also found that the RAYSI monomers are composed of the repetition of the same basic 120-bp repeat. Therefore, while the RAE730 monomers were built up by six repetitions of the 120-bp units (R730-1 to R730-6), the RAYSI monomers were formed by eight repetitions of the same unit (RAYSI-1 to RAYSI-8). An analysis of closeness between basic units of 120 bp reveals that both the RAE730 and the RAYSI satellite-DNA monomers have evolved from the basic 120-bp repeat unit through an intermediate trimer of 360 bp to a final duplication resulting in a 720-bp repeat unit (Fig. 2). The four intermediate 360-bp units differed less when belonging to the same satellite-DNA family, presumably indicating that the duplication from 360 to 720 bp occurred independently, at two different times, for each of the two satellite DNAs (Fig. 3). The RAYSI sequences underwent an additional length-amplification event by unequal crossing-over (Fig. 3).

A Sequence alignment between the two proposed 360-bp intermediates of the RAE730 satellite DNA (rae123 vs. rae456) in R. acetosa. Note that they shared 75% identity. The tree shows the relationships among the six 120-bp repeat units (R730-1 to R730-6) found within the RAE730 sequences, which support an intermediary step of 360 bp. B The relationships among the eight 120-bp repeat units forming the RAYSI sequences in R. acetosa (RAYSI-1 to RAYSI-8) are shown in the tree at the bottom. In this case, the partnership of the subrepeats by closeness is 1–6, 4–7, and 5–8 (see the alignment between the two proposed 360-bp intermediates, sharing 68% identity, of the RAYSI satellite DNA, –ray678 vs. ray145–). The two additional subrepeats, 2 and 3, are more similar to subrepeats 4 and 1, respectively. Thus, the origin of the RAYSI monomers could be explained as a consequence of two duplication events as for the RAE730 sequences followed by one additional amplification event by unequal crossing-over (see Fig. 3).

Schematic representation of the proposed mode of appearance of the current RAE730 and RAYSI satellite-DNA sequences from a basic repeat unit of 120 bp (1) through an intermediate satellite DNA of 360 bp (123), which duplicated to a 720-bp repeat unit (123456). The RAYSI sequences underwent an additional length-amplification event by unequal crossing-over (12123456 → 12345678).
In contrast, a comparative analysis of the different sequences revealed no evolutionary relationships between the RAE180 satellite-DNA sequences and the other two families, RAE730 and RAYSI.
Evolution of the Satellite-DNA Sequences of Rumex
Here we analyze the rate of concerted evolution of the three satellite-DNA families, for which we compare neighboring sequences (i.e., nonallelic monomers) in tandem arrays. For RAYSI, comparisons were made between sequences belonging to the subfamily RAYSI-J found in the two species, R. acetosa and R. papillaris. For all three satellite DNAs, we found similar variability levels within individuals and among individuals within a species. In the case of the RAE180 satellite DNA, where clones containing three or four adjacent monomers could be sequenced, variation levels between contiguous and noncontiguous monomers were similar. Table 1 summarizes the intraspecific and interspecific variation between monomers of each of the three satellite-DNA families of Rumex. In relation to interspecific divergence, the intraspecific variability of the Y-associated satellite DNAs, RAYSI-J and RAE180, was much higher than in the autosomic RAE730 sequences.
These results (Table 1) might indicate ancestral variation in RAYSI-J and RAE180 but not true divergence. Thus, we identified polymorphic sites shared between the two species compared for each satellite-DNA family. We assumed that these sites were ancestral and appeared prior to the split between R. acetosa and R. papillaris. In contrast, nonshared polymorphic sites are autapomorphies, representing different transitional stages in the process of intraspecific sequence homogenization and interspecific divergence. Therefore, shared polymorphic sites were excluded from the alignments, and genetic distances and the rates of sequence change were estimated from these alignments. This analysis is summarized in Table 2. Mean genetic distance for RAE730 sequences between R. acetosa and R. papillaris proved twofold to threefold higher than intraspecific variation. However, the difference between interspecific and intraspecific distances for RAYSI-J and RAE180 sequences was slight (0.7%). Table 2 also shows the evolutionary rates for each satellite-DNA family for a divergence time of 2 million years between R. acetosa and R. papillaris (Navajas-Pérez et al., unpublished data). The rate of sequence change for RAE730 satellite DNA was almost twofold higher than the rates for the RAYSI and the RAE180 satellite DNAs (Table 2).
A survey of the different transitional stages in the process of concerted evolution (Strachan et al. 1985) reveals that, while for RAE730 sequences a higher percentage of divergent sites are fully or almost fully homogenized sites (transition stages 4 and 5 according to Strachan et al. 1985), for RAYSI-J and RAE180 sequences most of the divergent polymorphic sites are initial stages in the process of sequence homogenization (stages 2 and 3), with few fully homogenized divergent sites (Table 2).
Discussion
Models of evolutionary dynamics for repetitive DNAs predict the preferential accumulation of tandem repetitive sequences in chromosomal regions with reduced levels of recombination and weak selective constraints on array size (Charlesworth et al. 1994). One characteristic region having these features is the differential segment of the Y chromosomes. The gradual suppression of recombination between the X and the Y chromosomes leads to the progressive degeneration of the Y by the loss of function of most genes (Filatov et al. 2000) and the expansion of tandem repetitive DNA families (Charlesworth 1996; Jobling and Tyler-Smith 2003). However, the processes that progressively degenerate the Y chromosome can be analyzed only in biological systems still under this evolutionary process (Charlesworth 2002). Thus, dioecious plant species are exceptional because there are a variety of species illustrating the different stages at which sex chromosomes are evolving (Charlesworth 2002; Ruiz Rejón 2004).
Rumex acetosa, in contrast to other dioecious plant species such as Silene latifolia, is useful for the study of the accumulation of satellite DNAs in the Y chromosomes because the Ys are heterochromatic and rich in satellite-DNA sequences (Ruiz Rejon et al. 1994; Shibata et al. 1999). However, though dioecy emerged at a similar time in Rumex and Silene (Navajas-Pérez et al., unpublished data), dioecious species of the latter genus have not accumulated repetitive DNA sequences in the Y chromosomes (Buzek et al. 1997; Scutt et al. 1997; Garrido-Ramos et al. 1999). Meanwhile, in the Rumex species analyzed here, Y degeneration was accelerated by the accumulation of RAYSI and RAE180 sequences. This acceleration presumably has been caused by the rearrangements giving rise to the multiple system of sex chromosomes (such as in R. acetosa and R. papillaris) since other species such as R. acetosella, with a simple system (XX/XY), have nondegenerated Y chromosomes as in S. latifolia (Löve 1944; Smith 1969).
Here, we present evidence indicating that RAYSI sequences are derived from an ancestral satellite DNA of 120 bp through an intermediate satellite DNA of 360 bp. Also, we found that this ancestral satellite led to RAE730, which has been amplified in a heterochromatic part of the genome outside the sex chromosomes. Specifically, this satellite DNA is the main constituent of the autosome supernumerary segments of R. acetosa (Shibata et al. 2000b) and R. papillaris (own observations). Thus, curiously, the same process, although twice independently, has given rise to two satellite-DNA families in two characteristic heterochromatic regions. Alternatively, it is feasible that the origin of the supernumerary segments and that of the Y chromosomes could be related. Common evolutionary pathways as well as common origins have been proposed at times for dispensable supernumerary DNA material (i.e., supernumerary chromosomes) and Y chromosomes (reviewed in Camacho et al. 2000).
We found low rates of evolution and sequence homogenization in the satellite DNAs in the Y chromosomes (RAYSI and RAE180) in relation to the autosomic RAE730 satellite DNA (Table 2). This difference is notable between RAYSI and RAE730 sequences since, although similar mechanisms produced the two satellite-DNA families, the two have evolved at different rates because of their different location. These data support the idea that there is no recombination between the Y chromosomes in Rumex and that this situation strongly influences the evolutionary pathways of satellite DNAs in sex chromosomes. Furthermore, the existence of two RAYSI subfamilies in R. acetosa could represent additional support for the contention that the two Y chromosomes do not recombine, since it has been proposed that mechanisms avoiding chromosomal interchanges, i.e., recombination, can lead to the formation of satellite DNA subfamilies (Pons and Gillespie 2003). In addition, sequence divergence between adjacent R180 repeats was similar to that between nonadjacent monomers. In contrast, if recombination were occurring, we would expect higher sequence-homogeneity levels between contiguous sequences than between distant sequences (Schueler et al. 2001).
Satellite DNAs evolve rapidly through concerted evolution (Dover 1982; Ugarkovic and Plohl 2002), sequence-change rates for satellite DNAs being higher than those of any other part of the eukaryotic genome (Ugarkovic and Plohl 2002). Different molecular mechanisms of DNA turnover, such as unequal crossing-over and gene conversion, are responsible for spreading new mutations throughout the members of the family at a rate of horizontal transference that is higher than the rate at which new changes arise (Dover 1982, 1986; Ohta and Dover 1984; Schlotterer and Tautz 1994). Thus, mechanisms avoiding chromosomal exchange (i.e., recombination) will negatively influence concerted evolution as expected for RAYSI and RAE180 sequences of the nonrecombining Y chromosomes. The rate of satellite-DNA evolution has been shown in the Drosophila obscura group to be 30 × 10−9 per site per year (Bachmann and Sperlich 1993), a high sequence-change rate as in the RAE730 sequences (22 × 10−9) in Rumex. This evolutionary rate of RAE730 sequences is approximately fourfold higher than for plant nonrepeating sequences (Gaut 1998). However, as hypothesized above, the RAYSI and RAE180 satellite-DNA sequences of Rumex species are evolving at a rate roughly half that of the RAE730 satellite DNA (Table 2). Thus, our data support the hypothesis that the satellite DNAs in the nonrecombining Y chromosomes show lower rates of sequence evolution than do autosomic satellite DNAs.
To estimate these rates, given the presence of many shared polymorphisms between the two species compared, we analyzed the sequences site by site, discarding the shared polymorphisms while keeping the polymorphisms that represent transition stages of differentiation. Sequence-evolution rates lower than those estimated with our method could be estimated from Da (Nei 1987), giving values twofold to fivefold higher for RAE730 sequences (14 × 10−9; Da = 0.056) than for RAE180 (7 × 10−9; Da = 0.028) and RAYSI-J (3 × 10−9; Da = 0.012) sequences. However, this parameter would not distinguish between ancestral and new polymorphisms and thus would disregard the transition stages of species differentiation. Our procedure assumes that shared polymorphisms are ancestral, although this assumption implies a certain underestimation of divergence (negligible compared to that of Da) since the procedure is unable to distinguish between ancestral polymorphisms and parallel mutations. However, we might expect a similar frequency of random parallel mutations for the three satellite-DNA families (i.e., a comparable underestimation of divergence), and therefore we might also expect proportionally the same differences in the evolutionary rate to persist between the three satellite-DNA families.
We found that within the RAE180 repeat units approximately 49% of the sites represented shared polymorphisms between R. acetosa and R. papillaris. However, we detected only three nearly fixed differences (1.5% of the sites) between these two species and 38% of polymorphic transitional stages. These data contrast with those found for the RAE730 sequences. In this case, 4% of nucleotide sites represented shared polymorphic sites, while 6.5% were fixed differences between R. acetosa and R. papillaris and 38% were transition differentiation stages. Clearly, the data support the contention that the rate of concerted evolution is lower for the RAE180 satellite DNA at the Y chromosomes than for the RAE730 autosomic satellite DNA, as it is for RAYSI sequences, since R. acetosa and R. papillaris differed by only 0.3% of the sites and showed 43% transitional stages. However, as opposed to RAE180, RAYSI sequences of the two species shared only 5% of polymorphisms. This difference in the number of shared polymorphisms could be explained by assuming that RAE180 sequences are older than RAYSI, and therefore have accumulated a higher number of ancestral polymorphisms. In fact, we are currently gathering data by means of Southern-blot hybridization indicating that RAE180 sequences have an older origin than do RAYSI sequences.
References
L Bachmann D Sperlich (1993) ArticleTitleGradual evolution of a specific satellite DNA family in Drosophila ambigua, D. tristis, and D. obscura Mol Biol Evol 10 647–659 Occurrence Handle1:CAS:528:DyaK3sXks1ymsLY%3D Occurrence Handle8336547
D Bachtrog (2003a) ArticleTitleAdaptation shapes patterns of genome evolution on sexual and asexual chromosomes in Drosophila Nat Genet 34 215–219 Occurrence Handle1:CAS:528:DC%2BD3sXktFSlt7g%3D
D Bachtrog (2003b) ArticleTitleAccumulation of Spock and Worf, two novel non-LTR retrotransposons, on the neo-Y chromosome of Drosophila miranda Mol Biol Evol 20 173–181 Occurrence Handle1:CAS:528:DC%2BD3sXhsVOitbo%3D
J Buzek H Koutníková A Houben K Ríha B Janousek J Siroky S Grant B Vyskot (1997) ArticleTitleIsolation and characterization of X chromosome-derived DNA sequences from a dioecious plant Melandrium album Chromosome Res 5 57–65 Occurrence Handle1:CAS:528:DyaK2sXivVyktr4%3D Occurrence Handle9088644
JP Camacho TF Sharbel LW Beukeboom (2000) ArticleTitleB-Chromosome evolution Philos Trans R Soc Lond Biol Sci 355 163–178 Occurrence Handle1:CAS:528:DC%2BD3cXjtVKqur8%3D
B Charlesworth (1996) ArticleTitleThe evolution of chromosomal sex determination and dosage compensation Curr Biol 6 149–162 Occurrence Handle1:CAS:528:DyaK28XhtFOjtLk%3D Occurrence Handle8673462
D Charlesworth (2002) ArticleTitlePlant sex determination and sex chromosomes Heredity 88 94–101 Occurrence Handle11932767
B Charlesworth P Sniegowski W Stephan (1994) ArticleTitleThe evolutionary dynamics of repetitive DNA in eukaryotes Nature 371 215– 220 Occurrence Handle10.1038/371215a0 Occurrence Handle1:CAS:528:DyaK2cXmt1emu7o%3D Occurrence Handle8078581
R De la Herrán F Robles N Cuñado JL Santos M Ruiz Rejón MA Garrido-Ramos C Ruiz Rejón (2001) ArticleTitleA heterochromatic satellite DNA is highly amplified in a single chromosome of Muscari (Hyacinthaceae) Chromosoma 110 197–202 Occurrence Handle11513294
G Dover (1982) ArticleTitleMolecular drive: A cohesive mode of species evolution Nature 299 111–117 Occurrence Handle10.1038/299111a0 Occurrence Handle1:CAS:528:DyaL3sXivFOjug%3D%3D Occurrence Handle7110332
G Dover (1986) ArticleTitleMolecular drive in multigene families: How biological novelties arise, spread and are assimilated Trends Genet 2 159–165 Occurrence Handle1:CAS:528:DyaL2sXltFWhsr8%3D
DA Filatov F Moneger I Negrutiu D Charlesworth (2000) ArticleTitleLow variability in a Y-linked plant gene and its implications for Y-chromosome evolution Nature 404 388–390 Occurrence Handle1:CAS:528:DC%2BD3cXitlyls7k%3D Occurrence Handle10746725
MA Garrido-Ramos R la Herrán Particlede M Ruiz Rejón C Ruiz Rejón (1999) ArticleTitleA subtelomeric satellite DNA family isolated from the genome of the dioecious plant Silene latifolia Genome 42 442–446 Occurrence Handle1:CAS:528:DyaK1MXkt1ChtbY%3D Occurrence Handle10382291
BS Gaut (1996) ArticleTitleMolecular clocks and nucleotide substitution rates in higher plants Evol Biol 30 93–120
MA Jobling C Tyler-Smith (2003) ArticleTitleThe human Y chromosome: An evolutionary marker comes of age Nat Rev Genet 4 598–612 Occurrence Handle1:CAS:528:DC%2BD3sXlvFaqt74%3D Occurrence Handle12897772
S Kumar K Tamura IB Jacobsen M Nei (2001) MEGA 2: Molecular Evolutionary Genetics Analysis software Arizona State University Tempe
A Löve (1944) ArticleTitleCytogenetic studies on Rumex subgenus acetosella Hereditas 30 1–136
A Löve (1967) ArticleTitleIOPB chromosome number reports XIII Taxon 16 448–454
M Nei (1987) Molecular evolutionary genetics Columbia Press New York
T Ohta G Dover (1984) ArticleTitleThe cohesive population genetics of molecular drive Genetics 108 501–521 Occurrence Handle1:CAS:528:DyaL2cXmt1Ght7k%3D Occurrence Handle6500260
J Pons RG Gillespie (2003) ArticleTitleCommon origin of the satellite DNAs of the Hawaiian spiders of the genus Tetragnatha: Evolutionary constraints on the length and nucleotide composition of the repeats Gene 313 169–177 Occurrence Handle1:CAS:528:DC%2BD3sXmvVart7c%3D Occurrence Handle12957388
J Pons E Petitpierre C Juan (2002) ArticleTitleEvolutionary dynamics of satellite DNA family PIM357 in species of the genus Pimelia (Tenebrionidae, Coleoptera) Mol Biol Evol 19 1329– 1340 Occurrence Handle1:CAS:528:DC%2BD38XmtF2rsrk%3D Occurrence Handle12140245
KH Rechinger SuffixJr (1964) The genus Rumex L TG Tutin VH Heywood NA Burges DH Valentine SM Walters DA Webb (Eds) Flora Europaea Vol I Cambridge University Press Cambridge 82–89
J Rozas R Rozas (1999) ArticleTitleDnaSP version 3: An integrated program for molecular population genetics and molecular evolution analysis Bioinformatics 15 174–175 Occurrence Handle10.1093/bioinformatics/15.2.174 Occurrence Handle1:CAS:528:DyaK1MXisVOksrY%3D Occurrence Handle10089204
C Ruiz Rejón M Jamilena MA Garrido-Ramos JS Parker M Ruiz Rejón (1994) ArticleTitleCytogenetic and molecular analysis of the multiple sex chromosome system of Rumex acetosa Heredity 72 209–215
M Ruiz Rejón (2004) Sex chromosomes in plants Encyclopedia of plant and crop science. Vol IV, Dekker Agropedia Marcel Dekker New York 1148–1151
N Saito M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic trees Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
C Schlotterer D Tautz (1994) ArticleTitleChromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution Curr Biol 4 777–783 Occurrence Handle10.1016/S0960-9822(00)00175-5 Occurrence Handle1:STN:280:ByqC38%2Fks1I%3D Occurrence Handle7820547
MG Schueler AW Higgins MK Rudd K Gustashaw HF Willard (2001) ArticleTitleGenomic and genetic definition of a functional human centromere Science 294 109–115 Occurrence Handle1:CAS:528:DC%2BD3MXnsFyitr4%3D Occurrence Handle11588252
CP Scutt Y Kamisugi F Sakai PM Gilmartin (1997) ArticleTitleLaser isolation of plant chromosomes: Studies on the DNA composition of the X and the Y sex chromosomes of Silene latifolia Genome 40 705–715 Occurrence Handle1:CAS:528:DyaK2sXntlSns7s%3D Occurrence Handle9352647
F Shibata M Hizume Y Kuroki (1999) ArticleTitleChromosome painting of Y chromosomes and isolation of a Y chromosome-specific repetitive sequence in the dioecious plant Rumex acetosa Chromosoma 108 266–270 Occurrence Handle1:CAS:528:DyaK1MXlt1Sjt74%3D Occurrence Handle10460415
F Shibata M Hizume Y Kuroki (2000a) ArticleTitleDifferentiation and the polymorphic nature of the Y chromosomes revealed by repetitive sequences in the dioecious plant, Rumex acetosa Chromosome Res 8 229–236 Occurrence Handle1:CAS:528:DC%2BD3cXjvVSjsrc%3D
F Shibata M Hizume Y Kuroki (2000b) ArticleTitleMolecular cytogenetic analysis of supernumerary heterochromatic segments in Rumex acetosa Genome 43 391–397 Occurrence Handle1:CAS:528:DC%2BD3cXjtVyjsrc%3D
H Skaletsky et al. (2003) ArticleTitleThe male-specific region of the human Y chromosome is a mosaic of discrete sequence classes Nature 423 825–837 Occurrence Handle1:CAS:528:DC%2BD3sXks1Kmtbk%3D Occurrence Handle12815422
BW Smith (1969) ArticleTitleEvolution of sex-determining mechanisms in Rumex Chromosomes Today 2 172–182
M Steinemann S Steinemann (1997) ArticleTitleThe enigma of the Y chromosome degeneration: TRAM, a novel retrotransposon is preferentially located on the neo-Y chromosome of Drosophila miranda Genetics 145 261–266 Occurrence Handle1:CAS:528:DyaK2sXntValu7g%3D Occurrence Handle9071582
T Strachan D Webb G Dover (1985) ArticleTitleTransition stages of molecular drive in multiple-copy DNA families in Drosophila EMBO J 4 1701–1708 Occurrence Handle1:CAS:528:DyaL2MXltFOnsrg%3D
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools Nucleic Acids Res 24 4876–4882 Occurrence Handle10.1093/nar/25.24.4876
D Ugarkovic M Plohl (2002) ArticleTitleVariation in satellite DNA profiles-causes and effects EMBO J 21 5955–5959 Occurrence Handle1:CAS:528:DC%2BD38XptFOrt7w%3D Occurrence Handle12426367
Acknowledgments
This work was supported by Grants BXX2000-1144-C02 and BOS2003-08737-C02-01 awarded by the Ministerio de Ciencia y Tecnología, Spain. We are deeply indebted to the Parque Nacional de Sierra Nevada and to the Parque Natural de la Sierra de Baza, both in Granada (Spain), for kindly providing the material analyzed in this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: DR. Jerzy Jurka]
Rights and permissions
About this article
Cite this article
Navajas-Pérez, R., la Herrán, R.d., Jamilena, M. et al. Reduced Rates of Sequence Evolution of Y-Linked Satellite DNA in Rumex (Polygonaceae). J Mol Evol 60, 391–399 (2005). https://doi.org/10.1007/s00239-004-0199-0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0199-0