
Abstract
The TFL1 and FT genes, which are key genes in the control of flowering time in Arabidopsis thaliana, belong to a small multigene family characterized by a specific phosphatidylethanolamine-binding protein domain, termed the PEBP gene family. Several PEBP genes are found in dicots and monocots, and act on the control of flowering time. We investigated the evolution of the PEBP gene family in cereals. First, taking advantage of the complete rice genome sequence and EST databases, we found 19 PEBP genes in this species, 6 of which were not previously described. Ten genes correspond to five pairs of paralogs mapped on known duplicated regions of the rice genome. Phylogenetic analysis of Arabidopsis and rice genes indicates that the PEBP gene family consists of three main homology classes (the so-called TFL1-LIKE, MFT-LIKE, and FT-LIKE subfamilies), in which gene duplication and/or loss occurred independently in Arabidopsis and rice. Second, phylogenetic analyses of genomic and EST sequences from five cereal species indicate that the three subfamilies of PEBP genes have been conserved in cereals. The tree structure suggests that the ancestral grass genome had at least two MFT-like genes, two TFL1-like genes, and eight FT-like genes. A phylogenomic approach leads to some hypotheses about conservation of gene function within the subfamilies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In higher plants, the timing of floral transition—the vegetative meristem’s turning into the reproductive state—is a major factor in plant adaptation. In Arabidopsis thaliana, an intricate network of signaling pathways controls this transition (Araki 2001; Koornneef et al. 1998; Mouradov et al. 2002). Two of the integrator genes, FT (FLOWERING LOCUS T) and TFL1 (TERMINAL FLOWER1), were identified by mutagenesis (Koornneef et al. 1991; Shannon and Meeks-Wagner 1991). Both genes encode very similar proteins almost exclusively made of a unique phosphatidylethanolamine-binding protein (PEBP) domain (domain accession: pfam01161). Despite their similarities, these genes have an opposite action on the flowering time: FT promotes flowering, while TFL1 delays it (Kobayashi et al. 1999). Together with four other closely related genes—TSF (TWIN SISTER OF FT), BFT (BROTHER OF FT AND TFL1), ATC (ARABIDOPSIS THALIANA CENTRORADIALIS HOMOLOGUE), and MFT (MOTHER OF FT AND TFL1; also known as E12A11)—they form the small PEBP family in Arabidopsis (Kardailsky et al. 1999; Kobayashi et al. 1999). PEBP genes have also been identified in animal systems. The molecular action of the PEBP proteins is not entirely clarified yet. Some studies support the hypothesis that they are involved in the regulation of a range of intracellular signaling cascades through their association with proteins of several functional classes. In mammals, they fix hydrophobic ligands, such as phosphatidylethanolamine, and nucleotides, like GTP (Banfield et al. 1998; Serre et al. 1998). Kroslak et al. (2001) report that human PEBP facilitates heterotrimeric G protein-coupled signaling.
TFL1-like genes have been found in various dicot species. In snapdragon, Antirrhinum majus, mutation in the CENTRORADIALIS (CEN) gene leads to the conversion of the indeterminate inflorescence architecture into a determinate one, by promoting a switch of the inflorescence meristem on a terminal symmetric flower (Bradley et al. 1996). The SELF PRUNING gene in tomato, Lycopersicum esculentum, controls the regularity of the floral transition along the compound shoot and therefore conditions the determinate vs. indeterminate growth habit of the plant (Carmel-Goren et al. 2003; Pnueli et al. 1998). The CET2/CET4 genes in tobacco, Nicotiana tabacum, are involved in the floral architecture and are expressed in vegetative meristems (Amaya et al. 1999). In pea, Pisum sativum, DETERMINATE acts to maintain the indeterminacy of the apical meristem during flowering and LATE FLOWERING (LF) delays the induction of flowering by prolonging the vegetative stage (Foucher et al. 2003). Allelic variation at the LF locus is an important component of natural variation for flowering time in pea. Therefore, the pathway influenced by TFL1-like genes may be an ancient and basic mechanism that controls flowering time and inflorescence architecture in dicot plants.
As in dicots, several PEBP genes have been identified in monocot species, namely, cereals. In rice, Oryza sativa L., the positional cloning of the major quantitative trait locus (QTL) for flowering time, Hd3 (Heading date3), led to the identification of two homologues of the Arabidopsis FT gene (Kojima et al. 2002). The search for orthologs of the TFL1 gene led to the identification of three new genes in rice, RCN1 (FRD2), RCN2, and RCN3 (FRD1) (Nakagawa et al. 2002). Izawa et al. (2002, 2003) used the almost-achieved sequencing of the subspecies indica rice genome to reveal that rice possesses at least 10 genes homologous to the FT gene. In perennial ryegrass, Jensen et al. (2001) have isolated a TFL1-like gene, LpTFL1, and characterized its role as a repressor of flowering time and as a controller of plant architecture. A recent study suggested that a homolog of Hd3a corresponds to a major QTL for heading date in ryegrass (Armstead et al. 2004). These results led to the assumption that the role of the PEBP gene family in the control of the flowering process could be conserved among cereals and, further, among monocots and dicots.
Like many species of agronomical interest (maize, wheat, barley, sorghum, etc.), rice belongs to the grass family, the Poaceae. It is the first cereal for which the genome sequence was released. However, for the main agronomic species, many expressed sequence tags (ESTs) derived from various tissue sample banks (stem, ear, leaf, grain, root, etc.) are available from public databases. In this study, first, we take advantage of the almost-complete sequencing of the rice genome (ssp. japonica) to search for the full repertoire of PEBP genes in this species and compare its complexity with the Arabidopsis repertoire. Second, we incorporate cereal EST and genomic sequences homologous to rice PEBP genes in a phylogenomic analysis (Eisen 1998), in order to obtain insight into the evolutionary history of the family and, eventually, infer possible functional conservation from in silico tissue-specific expression patterns.
Materials and Methods
Search for PEBP Sequences in Grasses
An extensive search of PEBP genes was conducted on rice genomic sequences. The sequences were obtained either from the annotation of the indica rice genomic sequences realized by Izawa et al. (2003) or by using Arabidopsis genes as query sequences in TBLASTX searches against the japonica rice BAC sequences. Then, in order to map them in silico, all the retrieved sequences were used as query in BALSTN searches against the japonica rice BAC sequences (available at http://www.gramene.org/). The genetic location of the BAC with the highest identity was identified.
The protein sequences of the six members of the PEBP family in Arabidopsis (FT, TFL1, TSF, ATC, BFT, and MFT) were used as query sequences for TBLASTN analysis of the EST contig databases of five grass species: rice, wheat, barley, maize, and sorghum. EST contigs are sequences of 5′ or 3′ parts of cDNAs and are thus incomplete sequences in nature. The ESTs extracted from the databases covered on average 60% of a typical PEBP coding sequence. Additionally, we searched for PEBP genes in maize and sorghum genomic sequences following the same query process. EST contig data and genomic sequences were obtained from the TIGR (http://www.tigr.org/tdb/tgi/plant.shtml) and PlantGDB (http://www.plantgdb.org/) databases, respectively. A ryegrass (Lolium perenne L.) sequence (GenBank accession number AF316419) which shows a high identity to the Arabidopsis TFL1 gene was included on the recovered sequence list
Phylogenetic Analysis
The complete alignment of PEBP sequences was manually edited using BioEdit 5.0.9 version (Hall 1991). Sequences were temporarily translated in order to delimit the 5′ and 3′ noncoding sequences of the ESTs. The parts located upstream of the ATG and downstream of the stop codon were discarded. Introns were removed from the genomic sequences. Only regions where the assessment of primary homology appeared reasonable were kept, generating a 594-nucleotide position matrix.
The phylogenetic relationships of nucleic sequences were investigated using neighbor-joining (NJ), maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI) methods. Any sites including gaps were discarded or considered as missing data, depending on the method used. The tree structure elaborated using the NJ method was based on the Jukes and Cantor gamma-corrected distance (α = 1.45, estimated using the ModelTest software [Posada and Crandall 1998]). NJ and MP methods were carried with the Mega2 software 3.0 (Kumar et al. 2001). Bootstraps with 1000 replicates were performed to assess node support in both analyses. For the ML tree, the (GTR + G + I, general time-reversible model estimating the proportion of invariable sites and gamma distribution) best-fitting models were selected using ModelTest 3.06 according to the Akaike Information Criterion (Posada and Crandall 1998). The ML phylogenetic analysis was performed with PAUP 4.10. BI was performed with MrBayes v3.0b4 (Ronquist and Huelsenbeck 2003), using a GTR model and site-specific rates partitioned by codon. In order to test the convergence of the system, one chain was run independently 10 times for 600,000 generations (burn-in period of 100,000 generations) sampled every 100 generations. Variance of each parameter estimated in each run was compared with variance of the average parameter calculated over the 10 runs. Then a single session was run for 50,100,000 generations (burn-in period of 100,000 generations) sampled every 100 generations. The Metropolis-coupled Markov chain Monte Carlo sampling approach was used to calculate posterior probabilities of clades. Phylogenetic analyses of translated sequences have been carried out and were congruent with results on the nucleotide matrix.
Results
Phylogenetic Analysis of Rice PEBP Genomic Sequences
Based on a genomewide analysis, we identified 19 PEBP genes in the genome of Oryza sativa ssp. japonica, of which 13 corresponded to genes previously described by Nakagawa et al. (2002) and Izawa et al. (2003) and 6 were new. Table 1 and Fig. 1 sum up the location of each rice genomic sequence revealed by in silico mapping. PEBP genes were dispersed on 7 of the 12 rice chromosomes. Among the 19 genes mapped, 10 (osFTL9/osFTL10, osFTL5/osFTL6, osFTL12/osFTL13, RCN2/RCN4, RCN1/RCN3) appear as five pairs belonging to duplicated chromosomal segments already identified by Paterson et al. (2003) and Salse et al. (2004). OsFTL2 and osFTL3 map at the same location on chromosome 6 (the two FT-like genes were present in the same BAC) and most probably were tandemly duplicated genes. The chromosomal segment bearing these two genes is duplicated at the end of chromosome 2, but no PEBP gene was mapped on this region.

Chromosomal localization of the PEBP family genes in Oryza sativa ssp. japonica. Centromeric region are drawn in gray. Black rectangles connected by dashed lines correspond to duplicated blocks (Paterson et al. 2003; Salse et al. 2004).
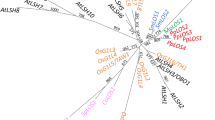
The evolutionary relationship between the 19 rice sequences and the 6 PEBP Arabidopsis sequences were investigated using NJ, ML, MP, and BI methods. The topologies of the NJ, MP, and ML trees and the 95% consensus tree from the Bayesian analysis were all congruent, except for a single node collapsing in the Bayesian consensus tree. We thus present only the result of the Bayesian analysis as an unrooted tree (Fig. 2). PEBP genes appear to be grouped in three well-supported clusters, each one associating Arabidopsis and rice sequences. Within each cluster, no clear orthology relationships emerge, suggesting independent evolution by gene duplication/loss within every species. Indeed, the first cluster, hereafter referred to as the MFT-LIKE subfamily, associates the Arabidopsis MFT gene and two rice genes, here called MFT1 and MFT2. The second cluster, the TFL1-LIKE subfamily, is composed of three Arabidopsis genes (TFL1, ATC, and BFT) and two groups of two rice genes, (RCN1, RCN3) and (RCN2, RCN4), found in duplicate chromosomal segments (see above). The RCN2/RCN4 group is not well supported, however, depending on the phylogenetic reconstruction method used. The last cluster, the so-called FT-LIKE subfamily, was composed of 2 Arabidopsis genes (FT and TSF) and 13 rice genes (osFTL1 to osFTL13). As in the TFL1-LIKE subfamily, several rice gene pairs are strongly associated and map in duplicate segments in the rice genome. FT and TSF Arabidopsis genes are closer to each other than to any other rice sequence, which is consistent with the hypothesis of duplication arising independently in rice and Arabidopsis.

Unrooted Bayesian tree of PEBP genes from rice Oryza sativa (os) and Arabidopisis thaliana (at). Support values for branches are shown and represent, from left to right, bootstrap values (1000 replicates) for NJ tree and MP consensus tree, and Bayesian frequencies (×100). Three major classes (TFL1-LIKE, MFT-LIKE, and FT-LIKE) are shown.
Phylogenetic Analysis of Cereal PEBP Sequences
Ninety-three coding sequences were found in the EST contig databases (47 sequences) and among the grass genomic sequences (46 sequences): 29 from rice, 29 from Triticeae (wheat, barley, and rye), 30 from maize, and 5 from sorghum (Table 2). Every genomic sequence has splicing sites at the same place as the Arabidopsis genes, as also observed with dicot PEBP genes (Amaya et al. 1999; Bradley et al. 1996; Carmel-Goren et al. 2003; Foucher et al. 2003; Pnueli et al. 1998). With three introns and four exons, the structure of the PEBP genes was conserved among cereals and Arabidopsis.
Phylogenetic analysis of all cereals and Arabidopsis PEBP sequences were performed using NJ, MP, ML, and BI methods. The tree topologies obtained using the NJ, MP, and BI methods were congruent, whereas the ML method did not provide a fully resolved tree with the full data set (data not shown). Bayesian analysis produced the most resolved tree presented in Fig. 3. First, one can notice that every rice EST contig is associated with one rice gene, suggesting a cognate origin. The lack of complete identity between a genomic sequence and cognate ESTs may come from sequencing errors or different genetic origins. In two cases, two or more ESTs were associated with one rice gene (Fig. 3). In the TFL1.2 group, TC158884 and AU093964 originate from the indica and japonica subspecies, respectively. In the FTL4 group, CB632234 and CA762716 are from the indica subspecies, while BM418838 originates from japonica subspecies. Moreover, the two indica ESTs correspond to either the 5′ or the 3′ part of a cDNA, which probably explains why they are not very close to each other. No rice EST contig was found unrelated to a genomic sequence, which strongly suggests that the complete repertoire of PEBP genes of rice is present in our genomic investigation. Second, the three subfamilies defined in the rice and Arabidopsis gene analysis are strongly supported (Bayesian support, 99%), consistent with the presence of three members of the PEBP family in the common ancestor of monocots and dicots.

Unrooted tree of the PEBP nucleotide sequences identified in cereals and Arabidopsis obtained by the Bayesian method. Bayesian posterior probabilities are given for branches. Major classes evidenced in previous analysis (TFL1-LIKE, MFT-LIKE, and FT-LIKE) and groups are shown (see text for details). Genomic sequence names are underlined. Abbreviations for species: Arabidopsis thaliana (at), Oryza sativa (os), Zea mays (zm), Sorghum bicolor (sb), Hordeum vulgare (hv), Triticum aestivum (ta), and Lolium perenne (lp).
The MFT-LIKE subfamily in grass consists of two homology groups, called MFT1 and MFT2. In each of them, sequences of the same species group first, then sequences from the same tribe. The most parsimonious hypothesis is that the grass common ancestor had two copies of the MFT-like gene (MFT1 and MFT2), and independent evolution proceeded in every species. In Triticeae, the two copies are represented by EST contigs of wheat and barley. Only the rice osMFT1 gene is associated with an EST contig. In maize, at least two genomic sequences are present in each group. Since these sequences come from the same genotype (inbred line B73), the polymorphism between sequences (SNPs) is caused by either sequencing errors or the presence of two paralogs. This last assumption is consistent with the known tetraploid origin of the maize genome (Gaut and Doebley 1997). The maize genomic sequences were associated with ESTs only in the MFT1 group.
Like the MFT-LIKE subfamily, the TFL1-LIKE subfamily is well structured, with the rice genes delimiting homology groups. The RCN3 and RCN1 genes associate within the TFL1.1 group, distantly related to the two other genes RCN2 and RCN4 (98% Bayesian values). Within the TFL1.1 group, sequences from sorghum and maize are close together (Panicoideae, 98% support) as well as sequences from wheat and ryegrass (Pooideae, 100% support). The relationship of these two sets with the two rice genes is not clear. By opposition to this group, we consider that the RCN2 and RCN4 genes form the second TFL1.2 group with most other grass sequences closer to RCN4 than to RCN2. The set of genes in cereals is not exhaustive, and the lack of homolog of RCN2 is thus inconclusive. The results could be accounted for by at least two TFL1-like genes in the cereal common ancestor, one corresponding to our TFL1.1 group, the other to the TFL1.2 group. It must be remembered that RCN1 and RCN3, on the one hand, and RCN2 and RCN4, on the other hand, mapped in duplicate genomic regions (chromosomes 1/12 and 2/4, respectively; see Fig. 1). Whether the duplications observed in rice predate the species divergence needs to be further investigated.
The FT-LIKE subfamily appears to be much more complex than the other two subfamilies, with eight well-supported homology groups associating rice genes and other cereal sequences (FTL1, FTL23, FTL910, FTL12, FTL13, FTL6, FTL4, and FTL7). In most of them, grass sequences are grouped by species, then by tribe or subfamily. Keeping in mind the nonexhaustivity of the PEBP data set in cereals, it can be suggested that the grass common ancestor had at least eight FT-like genes. FTL12 and FTL13 correspond to homology groups identified by two rice sequences mapped on duplicate genomic regions, suggesting that this specific duplication predates grass divergence. The same hypothesis could be formulated within the FTL910 group, albeit the divergence between the two sets of sequences associated with the paralogues FTL10 and FTL9 is not well supported. Within group FTL23, rice osFTL2 (Hd3a) and osFTL3 are very close to each other, and associated with wheat and maize EST contigs. The topology supports the rice mapping data (both genes are present in the same BAC on chromosome 6), suggesting a recent tandem duplication, which would be rice specific. Three rice genes (osFTL5, osFTL8, and osFTL11) are not associated with other cereal sequences, suggesting that they might be specific to rice.
In several cases, more than one wheat sequence belongs to the same homology group. This can be explained either by sequencing errors on the ESTs, by different genetic origins (numerous varieties are used to build the wheat databases), or by the expression of different homeologous genes carried by the three A, B, and D wheat genomes.
Discussion
The initiation of flowering is modulated by both environmental and endogenous signals, such as photoperiod, vernalization, and gibberellic acid. Molecular genetic studies have revealed that homologous genes in rice, a short-day plant, and Arabidopsis thaliana, a long-day plant, such as Heading date 1 (Hd1)/CONSTANS (CO), Hd3a/FLOWERING LOCUS T (FT), and osSOC1/SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), are implicated in the regulation of flowering time (Kojima et al. 2002; Tadege et al. 2003; Yano et al. 2000). However, conservation of these genes between rice and Arabidopsis does not necessarily imply conservation of gene function. Indeed, while the promotion of flowering in long days in Arabidopsis results from CO activating FT, the delay in flowering in long days in rice results from Hd1 repressing Hd3a (Izawa et al. 2002).
FT gene is a member of a small gene family, the PEBP gene family, which includes TFL1, ATC, BFT, MFT, and TSF in Arabidopsis (Kobayashi et al. 1999; Mimida et al. 2001). In this plant, most members of the PEBP family act as regulators of flowering time: TFL1 delays flowering time and constitutive expression of TSF and ATC, and MFT causes early flowering. Several PEBP genes have been identified in rice (Izawa et al. 2003; Nakagawa et al. 2002), some of which modulate flowering time: overexpression of RCN1 and RCN2, the rice homologs of Arabidopsis TFL1, leads to delay in flowering (Nakagawa et al. 2002), whereas ectopic expression of Hd3a (osFTL2), RFT1 (osFTL3), and FTL (osFTL1) results in early flowering phenotypes (Izawa et al. 2002; Kojima et al. 2002). It is thus probable that the family members constitute a signaling mediator to determine flowering time in dicots as well as in monocots.
In order to gain insight into the evolutionary history of the PEBP gene family in grasses and to infer the role of some of its members in the flowering process, we first compare the repertoire of PEBP genes in Arabidopsis and rice. Combining data from the almost-sequenced rice genome and ESTs in databases, we retrieved 19 genes from the PEBP family, which likely reflect the full repertoire of this gene family in rice. Thus, the family is much more complex in this species than in the eudicot Arabidopsis (Izawa et al. 2002). The phylogenetic analysis of the sequences of the two species allows three subfamilies to be identified. Among these, the FT-LIKE subfamily appears to be the largest in rice, with 13 members, while the TFL1-LIKE subfamily could be considered the most complex in Arabidopsis, with 3 members. Within each subfamily no clear orthology relationships emerge between Arabidopsis and rice genes, indicating independent evolution by duplication (or gene loss) in the two species. In rice, the multiplicity of paralogues in the TFL1-like and FT-like subfamilies originates at least in part from duplication of chromosomal regions (Paterson et al. 2003; Salse et al. 2004; Vandepoele et al. 2003). Vandepoele et al. (2003) suggested that the duplication of the rice genome predates the divergence of most grasses. Although the nodes have low support, the homology groups comprising osFTL12 and osFT13, mapped in duplicate segments of chromosome 6 and chromosome 2, respectively, and those comprising osFTL10 and osFT9, mapped in duplicate segments of chromosome 1 and chromosome 5, respectively, sustain this hypothesis. On the other hand, functional redundancy could lead to a loss of one duplicate. Such a process might be responsible for the lack of FT-like genes in the segment of chromosome 2 corresponding to a duplication of a segment of chromosome 6 bearing the pair of tandemly duplicated genes osFTL2 and osFTL3.
In the second step, a phylogenetic approach was applied to all PEBP sequences found in EST contigs and genomic sequences databases for six cereal species and incorporating the six PEBP genes of Arabidopsis. As previously noted by Citerne et al. (2003), phylogenetic reconstruction using BI gives a more fully resolved tree than the parsimony method. The topology of the tree confirms the organization of the PEBP sequences in three subfamilies, whose complexity is higher in cereals than in Arabidopsis. The three subfamilies are unequally represented according to the species, most probably because some of them have been less fully investigated than others (2:4:13 in rice, 7:4:9 in wheat, 5:0:4 in barley, 7:7:16 in maize, and 2:2:1 in sorghum for relative gene number of the MFT-LIKE, TFL1-LIKE, FT-LIKE subfamilies, respectively). The definition of homology groups in relation to one reference, rice, facilitates the annotation of new and/or incomplete sequences such as ESTs. Moreover, it allows several hypotheses about the evolutionary history of the PEBP gene family in cereals to be proposed. Thus, based on the structure of the homology groups associating at least one rice sequence and at least one other cereal sequence, the most parsimonious hypothesis suggests that two MFT-like and two TFL1-like genes and at least eight FT-like genes were present in the ancestral grass genome (see Fig. 3). Subsequently, these genes likely evolved independently in each taxon by duplication and possibly gene loss, thus often confusing orthology relationships within the subfamilies.
This multiplicity of family members raises questions about the functional diversification and conservation within the PEBP family in cereals. Conservation of expression patterns among homologous genes strongly suggests functional conservation. The expression patterns of known genes (e.g., FT, TFL1, Hd3a) allows hypotheses about the function of cereal genes of each homology groups to be proposed. Moreover, the nature of the plant sample that was used to build the cereal EST databases may also provide some preliminary trends about gene expression (in silico expression; Table 3). However, data on the quality and depth of these databases are limited, and no information is available as to when the tissue samples were harvested during the day. Thus the absence of an EST in a database does not prove lack of gene expression within the corresponding tissue or organ, and specificity of expression cannot be established from these data. In situ expression analyses and/or RT-PCR would be required to refine data from in silico expression.
The MFT-LIKE subfamily associates the MFT gene of Arabidopsis and two homology groups in cereals. Little is known about the role of MFT gene in Arabidopsis. In a recent study, Yoo et al. (2004) found that overexpression of MFT accelerates flowering time but loss of function of MFT did not show any obvious phenotype. The authors suggested that MFT functions as a floral inducer and may act redundantly in determination of flowering time in Arabidopsis. In silico analyses showed that MFT-like genes in cereals are expressed in grain or spike after pollination, and no differentiation was apparent between MFT1 and MFT2 homology groups. These results suggest that MFT-like genes could play a role in the grain maturation process rather than in the flowering process in cereals.
The organization of the TFL1-LIKE subfamily suggests at least two homology groups, each one comprising a pair of rice genes most probably originating from a chromosomal duplication. Nakagawa et al. (2002) found the rice RCN3/FRD1 gene to be chimeric, suggesting that it was nonfunctional. However, our analysis shows that the RCN3 indica rice gene is not chimeric and a putative cognate EST (TC143228) is expressed in leaf (see Table 3). RCN1 (chromosome 11) and RCN2 (chromosome 2) genes are expressed in the meristem and have an action quite similar to that of the TFL1 gene in Arabidopsis when overexpressed (Nakagawa et al. 2002), namely, a flowering delay with a repression of the floral transition. Since none of these genes is a true ortholog of TFL1, this suggests either an ancestral function in the flowering process that was conserved among some TFL1-like genes or, alternatively, independent recruitment for a similar function of different genes from the same PEBP subfamily. The ATC gene in Arabidopsis that also belongs to the TFL1-LIKE subfamily has a quite different expression pattern, being expressed in the hypocotyls of young plants but not in the meristem (Mimida et al. 2001). Cereal ESTs of the TFL1-LIKE subfamily are found preferentially in the inflorescence, suggesting that at least some TFL1-like genes may have an action during flowering that could be similar to that of Arabidopsis TFL1. However, conservation of gene function and downstream pathways remains to be established.
Within the FT-LIKE subfamily, cognate ESTs were not found for all rice genes, which raises the question of their functionality, particularly for osFTL7, osFTL8, osFTL9, osFTL10, osFTL11, osFTL12, and osFTL13. The osFTL1 to osFTL9 genes were shown to be expressed in leaves (Doi et al. 2004; Izawa et al. 2003). Moreover, it was recently shown that Ehd1, a gene involved in short-day promotion of flowering can specifically induce FT-like genes osFTL1, osFTL2, osFTL3, and osFTL9 in a Hd1-deficient background (Doi et al. 2004). ESTs coming from other cereal species are present in the FTL1, FTL23, FTL10, FTL12, and FTL13 homology groups. It can be noticed that osFTL11 maps very close to the centromere on chromosome 11, which may suppress expression. Functionality of most FT-like genes is reinforced by the fact that we never found a frameshift or stop codon which would alter transcription or protein functionality. osFTL2 has been characterized as a QTL of flowering time in rice (Kojima et al. 2002). This gene has homologs in maize and wheat. The in silico expression analysis of cereal ESTs in homology group FTL23 is consistent with the reported expression of Hd3a in rice (Kojima et al. 2002) and of the FT gene in Arabidopsis, showing the main expression in the stem and the leaves (Kobayashi et al. 1999). A recent study shows that a heading-date QTL in ryegrass (Lolium perenne L.) seems to be the syntenous region of the Hd3a locus in rice (Armstead et al. 2004). It would therefore be very likely that the gene has a conserved function in wheat and maize. Confirming this assumption would require (i) mapping the genes homologous to Hd3a in maize and wheat, (ii) comparing the map location to the QTL affecting flowering time in these species, and (iii) conducting association tests between allelic forms and quantitative variation in photoperiod response in a population. It would also help to localize the essential sites for gene action and compare these sites between species.
Several models have been proposed to account for the persistence of duplicated genes over long evolutionary periods. Indeed, strict functional redundancy is not expected over time. Several potential fates may be experienced by duplicated genes, namely, subfunctionalization, neofunctionalization, and degeneracy through accumulation of deleterious mutations leading to pseudogenes (Lynch and Force 2000; Ohno 1970, 2000). Examples of subfunctionalization have recently been described in allopolyploid coton and Cycloidea-like genes in Antirrhineae (Adams et al. 2003; Hileman and Baum 2003). The PEBP genes consist of a single highly conserved domain which represents more that 80% of the coding sequence. Possible subfunctionalization within this gene family would more likely concern cis-regulatory sequences, leading to various temporal and/or tissue-specific expressions. Several arguments are consistent with this hypothesis. Two haplotypes of the TFL1 promoter seem to be maintained by selection in Arabidopsis, while low-frequency polymorphisms were observed in its coding region (Olsen et al. 2002). The coding sequence of Hd3a is almost fully conserved between the two rice cultivars Nipponbare and Kasalath, the parental lines of the segregating population where osFTL2 was found as a QTL of flowering date (Kojima et al. 2002). Analysis of promoter sequences in rice and other grasses would contribute to a better understanding of functional divergence within the PEBP family and subfamilies.
References
Adams KL, Cronn R, Percifield R, Wendel JF (2003) Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing Proc Natl Acad Sci USA 100:4649–4654
Amaya I, Ratcliffe OJ, Bradley DJ (1999) Expression of CENTRORADIALIS (CEN) and CEN-like genes in tobacco reveals a conserved mechanism controlling phase change in diverse species Plant Cell 11:1405–1418
Araki T (2001) Transition from vegetative to reproductive phase Curr Opin Plant Biol 4:63–68
Armstead IP, Turner LB, Farrell M, Skot L, Gomez P, Montoya T, Donnison IS, King IP, Humphreys MO (2004) Synteny between a major heading-date QTL in perennial ryegrass (Lolium perenne L.) and the Hd3 heading-date locus in rice Theor Appl Genet 108:822–828
Banfield MJ, Barker JJ, Perry AC, Brady RL (1998) Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction Structure 6:1245–1254
Bradley D, Carpenter R, Copsey L, Vincent C, Rothstein S, Coen E (1996) Control of inflorescence architecture in Antirrhinum Nature 379:791–797
Carmel-Goren L, Liu YS, Lifschitz E, Zamir D (2003) The SELF-PRUNING gene family in tomato Plant Mol Biol 52:1215–1222
Citerne HL, Luo D, Pennington RT, Coen E, Cronk QCB (2003) A phylogenomic investigation of CYCLOIDEA-like TCP genes in the Leguminosae Plant Physiol. 131:1042–1053
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18:926–936
Eisen JA (1998) Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis Genome Res 8:163–167
Foucher F, Morin J, Courtiade J, Cadioux S, Ellis N, Banfield MJ, Rameau C (2003) DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER1/CENTRORADIALIS homologues that control two distinct phases of flowering initiation and development in pea Plant Cell 15:2742–2754
Gaut BS, Doebley JF (1997) DNA sequence evidence for the segmental allotetraploid origin of maize Proc Natl Acad Sci USA 94:6809–6814
Hall TA (1991) BioEdit: A user-friendly biological sequence alignment editor and analysis. Ibis Theropeutics, Carlsbad, CA
Hileman LC, Baum DA (2003) Why do paralogs persist? Molecular evolution of CYCLOIDEA and related floral symmetry genes in Antirrhineae (Veronicaceae) Mol Biol Evol 20:591–600
Izawa T, Oikawa T, Sugiyama N, Tanisaka T, Yano M, Shimamoto K (2002) Phytochrome mediates the external light signal to repress FT orthologues in photoperiodic flowering of rice Genes Dev 16:2006–2020
Izawa T, Takahashi Y, Yano M (2003) Comparative biology comes into bloom: genomic and genetic comparison of flowering pathways in rice and Arabidopsis Curr Opin Plant Biol 6:113–120
Jensen CS, Salchert K, Nielsen KK (2001) A terminal Flower1–like gene from perennial ryegrass involved in floral transition and axillary meristem identity Plant Physiol 125:1517–1528
Kardailsky I, Shukla VK, Ahn JH, Dagenais N, Christensen SK, Nguyen JT, Chory J, Harrison MJ, Weigel D (1999) Activation tagging of the floral inducer FT Science 286:1962–1965
Kobayashi Y, Kaya H, Goto K, Iwabuchi M, Araki T (1999) A pair of related genes with antagonistic roles in mediating flowering signals Science 286:1960–1962
Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M (2002) Hd3a, a rice orthologue of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions Plant Cell Physiol 43:1096–1105
Koornneef M, Alonso–Blanco C, Peeters AJ, Soppe W (1998) Genetic control of flowering time in Arabidopsis Annu Rev Plant Physiol Plant Mol Biol 49:345–370
Koornneef M, Hanhart CJ, Van der Veen JH (1991) A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana Mol Gen Genet 229:57–66
Kroslak T, Koch T, Kahl E, Hollt V (2001) Human phosphatidylethanolamine–binding protein facilitates heterotrimeric g protein-dependent signaling J Biol Chem 276:39772–39778
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: Molecular Evolutionary Genetics Analysis software. Arizona State University, Tempe
Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization Genet Soc Am 154:459–473
Mimida N, Goto K, Kobayashi Y, Araki T, Ahn JH, Weigel D, Murata M, Motoyoshi F, Sakamoto W (2001) Functional divergence of the TFL1–like gene family in Arabidopsis revealed by characterization of a novel homologueue Genes Cells 6:327–336
Mouradov A, Cremer F, Coupland G (2002) Control of flowering time: interacting pathways as a basis for diversity Plant Cell 14(Suppl): S111–S130
Nakagawa M, Shimamoto K, Kyozuka J (2002) Overexpression of RCN1 and RCN2, rice TERMINAL FLOWER 1/CENTRORADIALIS homologues, confers delay of phase transition and altered panicle morphology in rice Plant J 29:743–750
Ohno (1970) Evolution by gene duplication Springer-Verlag, Berlin
Ohta T (2000) Evolution of gene families Gene 259:45–52
Olsen KM, Womack A, Garrett AR, Suddith JI, Purugganan MD (2002) Contrasting evolutionary forces in the Arabidopsis thaliana floral developmental pathway Genetics 160:1641–1650
Paterson AH, Bowers JE, Peterson DG, Estill JC, Chapman BA (2003) Structure and evolution of cereal genomes Curr Opin Genet Dev 13:644–650
Pnueli L, Carmel-Goren L, Hareven D, Gutfinger T, Alvarez J, Ganal M, Zamir D, Lifschitz E (1998) The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the orthologue of CEN and TFL1 Development 125:1979–1989
Posada D, Crandall K (1998) MODELTEST: testing the model of DNA substitution Bioinformatics 14:817–818
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models Bioinformatics 19:1572–1574
Salse J, Piegu B, Cooke R, Delseny M (2004) New in silico insight into the synteny between rice (Oryza sativa L.) and maize (Zea mays L.) highlights reshuffling and identifies new duplications in the rice genome Plant J 38:396–409
Serre L, Vallee B, Bureaud N, Schoentgen F, Zelwer C (1998) Crystal structure of the phosphatidylethanolamine-binding protein from bovine brain: a novel structural class of phospholipid-binding proteins. Structure 6:1255–1265
Shannon S, Meeks-Wagner DR (1991) A mutation in the Arabidopsis TFL1 gene affects inflorescence meristem development Plant Cell 3:877–892
Tadege M, Sheldon CC, Helliwell CA, Upadhyaya NM, Dennis ES, Peacock WJ (2003) Reciprocal control of flowering time by OsSOC1 in transgenic Arabidopsis and by FLC in transgenic rice Null 1:361–369
Vandepoele K, Simillion C, Van de Peer Y (2003) Evidence that rice and other cereals are ancient aneuploids Plant Cell 15:2192–2202
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T (2000) Hd1, a major photoperiod sensitivity QTL in rice, is closely related to the Arabidopsis flowering time gene CONSTANS Plant Cell 12:2473–2483
Yoo SY, Kardailsky I, Lee JS, Weigel D, Ahn JH (2004) Acceleration of flowering by overexpression of MFT (MOTHER OF FT AND TFL1) Mol Cells 17:95–101
Acknowledgments
We are grateful to Domenica Manicacci, Maud Tenaillon, and Alain Charcosset for critical reading of the manuscript. This research was supported by a grant to Fabien Chardon from the Génoplante programme.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Yves Van de Peer]
Rights and permissions
About this article
Cite this article
Chardon, F., Damerval, C. Phylogenomic Analysis of the PEBP Gene Family in Cereals. J Mol Evol 61, 579–590 (2005). https://doi.org/10.1007/s00239-004-0179-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0179-4