
Abstract
Using a phylogenetic approach, we discovered three putative horizontal transfers between bacterial and archaeal species involving large clusters of genes. One transfer involves an operon of 13 genes, called mbx, wich probably was transferred into the genome of Thermotoga maritima from a species belonging or close to the Pyrococcus genus. The two others implied an operon of six genes, called ech, transferred independently to the genomes of Thermoanaerobacter tengcongensis and Desulfovibrio gigas, from a species belonging or close to the Methanosarcina genus. All these transfers affected operons coding for multisubunit membrane-bound (NiFe) hydrogenases involved in the energy metabolism of the donor genomes. The functionality of the transferred operons has not been experimentally demonstrated for T. maritima, whereas in D. gigas and T. tengcongensis the encoded multisubunit hydrogenase could have a role in energy conservation. This report adds several cases of horizontal gene transfers among hydrogenases already described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the early work of Médigue et al. (1991) in Escherichia coli, horizontal gene transfer (HGT) is known to be a major force in the evolution of prokaryotic genomes (Jain et al. 2003). Different types of methods have been proposed to detect HGT events from genome sequence data. One way to detect HGTs is based on univariate or multivariate codon or oligonucleotide usage measures (for some examples see Médigue et al. 1991; Lawrence and Ochman 1998; Karlin et al. 1998ab; Garcia-Vallvé et al. 2000; Nicolas et al. 2002). Notably, it was the use of such methods that led to the idea that HGTs are extremely frequent in the prokaryotic world. Many critics arose about the limitations of these methods, especially showing that they tend to overestimate the number of transfers (Koski et al. 2001; Wang 2001; Guindon and Perrière 2001; Daubin and Perrière 2003).
The hypothesis of HGT can also be based on results of BLAST (Altschul et al. 1997) database searches when a sequence from a distantly related organism is high on the list of matches. This approach has been used to demonstrate that HGTs happened between hyperthermophilic bacteria and archaea (Aravind et al. 1998; Nelson et al. 1999), between eukaryotic organisms and Mycobacterium tuberculosis (Gamielden et al. 2002), and even between human and bacterial species (Lander et al. 2001). Some of them have been confirmed by experimental studies (Nesbo et al. 2001) but many have been dismissed later, mainly by the use of phylogenetic methods (Kyrpides and Olsen 1999; Koski and Golding 2001; Stanhope et al. 2001; Kinsella and McInerney 2003). Therefore, it seems that the most accurate way to study HGTs events to date is the construction and the analysis of phylogenetic trees.
The different methods presented above have been used to estimate the amount of transferred genes into the genomes of the hyperthermophilic bacteria Thermotoga maritima and Aquifex aeolicus. Analyses based on BLAST similarities of the complete genomes of A. aeolicus (Deckert et al. 1998) and T. maritima (Nelson et al. 1999) revealed that, respectively, 16 and 24% of their coding sequences were most similar to genes of archaea than to genes of bacteria. Some authors thought that these observations were evidence of massive HGTs between archaeal and bacterial hyperthermophiles (Aravind et al. 1998; Nelson et al. 1999). A phylogenomic approach has also been used by Sicheritz-Pontén and Andersson (2001); they have estimated the phylogenetic connections between different species of bacteria and archaea and shown that 16–26% of the genes of A. aeolicus and T. maritima had pure archaeal connections, suggesting that these genes could have arosen in their genomes by HGT. In the case of T. maritima, experiments on subtractive hybridization have shown that there was a great variability of genome content between close strains of Thermotoga and that it could be attributed to HGT events (Nesbo et al. 2002).
As the case of HGTs between hyperthermophilic bacteria and archaea is still a hot topic (Makarova et al. 2003), we decided to make phylogenetic analyses on theses species using a special release of HOBACGEN, a gene family database devoted to prokaryotes (Perrière et al. 2000). This release, called HOBACGEN-CG, has been established with 86 prokaryotes (70 bacteria and 16 archaea) for which the complete genome sequence was available. Particularly, we focused on the families containing sequences of the three completely sequenced hyperthermophilic bacteria: T. maritima, A. aeolicus, and Thermoanaerobacter tengcongensis. The latter has an optimal growth temperature (Topt) that is lower than 80°C, the lower limit commonly used to define hyperthermophilic organisms, but it can grow at 80°C. Moreover, this bacterium possesses the gene coding for reverse gyrase, which is the only specific gene common to all hyperthermophilic bacteria and archaea (Forterre 2002). So we considered this organism as a hyperthermophilic bacterium.
We detected three large HGTs events implying entire operon structures that happened between bacterial and archaeal species. These transfers implied two different operons coding for multisubunit transmembrane complexes involved in energetic metabolism. The first one is related to mbx, an operon of 13 genes which probably was transferred into the genome of T. maritima by horizontal transfer from an archaea belonging to the Pyrococcus group. The two others are related to ech, a six-gene operon which has probably been transferred independently into the genomes of two bacteria, T. tengcongensis and Desulfovibrio gigas, from an archaea belonging to the Methanosarcina clade.
Materials and Methods
Homologous Gene Families
The HOBACGEN-CG database was created with the protein sequences from 86 prokaryotes (70 bacteria and 16 archaea) for which the complete genome sequence was available. When more than one strain was available for a given species, only the type strain was selected (e.g., K12 in the case of E. coli). All these sequences were extracted from the SWISS-PROT/TrEMBL collection (Boeckmann et al. 2003) and then clustered into families of homologous genes using the procedure described by Perrière et al. (2000). Once the database was established, it was possible to select the families that contained at least one sequence of the three hyperthermophilic bacteria and sequences of archaea. We then looked at the trees corresponding to each family, and we retained those where a hyperthermophilic bacterium was grouped with archaeal species. For the different HOBACGEN-CG families that correspond to the genes belonging to the two operons studied here, we completed the original data with other homologs (not necessarily belonging to completely sequenced organisms) found by similarity in SWISS-PROT/TrEMBL. This allowed us to introduce a broader set of species in the phylogenetic trees computed.
Alignments and Trees
The sequences of each family were realigned using CLUSTAL W (Higgins et al. 1996), with all the default parameters. Then we filtered the alignments using the program GBLOCKS (Castresana 2000) in order to select only well-aligned parts. This program identifies blocks in an alignment for which homology of sites can be assumed with good confidence, and regions that contain reliable phylogenetic information. When the filtering by GBLOCKS was too stringent or when the sequences were too short, we corrected the alignments manually.
For each family, we computed two trees: one with BIONJ (Gascuel 1997) and one with PHYML, a fast and efficient maximum likelihood (ML) method (Guindon and Gascuel 2003). For both methods, we used the Jones–Taylor–Thornton (JTT) model of amino acid substitution (Jones et al. 1992) and 1000 bootstrap replicates were made, using SEQBOOT from the PHYLIP package (Felsenstein 1989) and a program developed by us; ADDBOOTSTRAP. In the case of BIONJ trees, the distances were computed with PROTDIST from PHYLIP. Heterogeneities between sites were estimated under a gamma law-based model of substitution, and the computation of the alpha parameter was carried out by TREE-PUZZLE (Strimmer and von Haeseler 1996). For ML trees, heterogeneities between sites were also estimated under a gamma law based model of substitution, with estimation of the alpha parameter by PHYML. All computations were made on a multiprocessor Linux cluster containing more than 600 CPUs.
Adjacent Regions
Each time a phylogenetic incongruency (i.e., at least one sequence of a hyperthermophilic bacterium was clustered within an archaeal clade) supported by a high bootstrap value was detected in a gene family, we analyzed the adjacent sequences on the chromosome. For that purpose, we searched to which family of HOBACGEN-CG adjacent genes belonged, and whenever possible, we recalculated their phylogeny using the method described above. We extended the family data with other homologs found in non–completely sequenced organisms. More precisely, we selected only those that were more similar to homologs from hyperthermophilic bacteria than from completely sequenced organisms with which the hyperthermophilic bacteria were grouped in the original phylogeny.
Availability
All the data used in this study (complete list of sequences used in the analyses, multiple alignments, and trees) can be downloaded from the Pôle Bioinformatique Lyonnais (PBIL) Web server at http://pbil.univ-lyon1.fr/datasets/Calteau2004. The HOBACGEN-CG database can be accessed through the PBIL query interface at http://pbil.univ-lyon1.fr/search/query_fam.php. The ADDBOOTSTRAP program is distributed upon request by Manolo Gouy (mgouy@biomserv.univ-lyon1.fr). This program has been written in standard ANSI C and is compatible with the PHYLIP package.
Results
Among the families we studied, two of them presented remarkable features in terms of possible HGTs in hyperthermophilic bacteria and permitted the detection of the different operon transfers. These two families are annotated as subunits of the NADH:ubiquinone oxidoreductase complex, and their accession numbers in HOBACGEN-CG are HBG000146 and HBG003659. The topologies of the ML and BIONJ trees for members of these families and for their neighbors on the chromosome were always congruent relatively to the position of the species for which we detected putative HGTs. Therefore, only the ML trees have been represented in the following section to support the results.
HGT of a 13-Gene Cluster in T. maritima
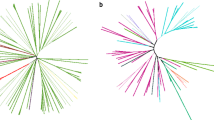
The phylogenetic trees of the two families mentioned above showed incongruencies related to the position of sequences of T. maritima (Fig. 1). They were grouped with high bootstrap support with the sequences of three archaea belonging to the Pyrococcus genus (P. abyssi, P. furiosus, and P. horikoshii). As these two genes are neighbors on the chromosome of T. maritima, we analyzed the genes in their neighborhood, and we found that a total of 12 consecutive genes (including these 2) were more similar to sequences of Pyrococcus than to bacterial sequences. Moreover, the order of all these genes is conserved between T. maritima and Pyrococcus. For eight of them, the grouping of T. maritima with Pyrococcus species in the corresponding tree was supported by a high bootstrap value (>80%). For the four other genes, the bootstrap values were low or we could not make a tree due to the lack of a sufficient number of detected homologs.

Maximum-likelihood trees of the HBG000146 (A) and HBG003659 (B) families from HOBACGEN-CG. Bacterial species are in boldface and the arrows indicate the location of the three bacteria (T. tengcongensis, D. gigas, and T. maritima) for which an HGT is suspected. Only bootstrap values over 50% are shown for the internal branches.
The genes of the three Pyrococcus species are annotated as hypothetical proteins or NADH dehydrogenases in SWISS-PROT/TrEMBL (Table 1). In P. furiosus they are known to belong to the mbx operon (Fig. 2), while in the other species they are not attached to any structure or organisation of that kind. This operon contains 13 genes, which code for a putative energy transducing membrane complex (Sapra et al. 2000; Silva et al. 2000), and homologous operons exist in P. abyssi and P. horikoshii. As we detected only 12 corresponding genes in T. maritima, we studied more closely the gene located immediately after the cluster. This gene (TM1217) codes for a glutamate synthase and belongs to a family of HOBACGEN-CG which contains no sequences of Pyrococcus. The BLAST search, using the P. furiosus gene as a query sequence, finds the two homologs in P. abyssi and P. horikoshii and the third hit corresponds to the gene TM1217, but the similarity is limited to its first 120 amino acids. This suggests that the transfer event consisted in an insertion of the whole mbx operon in this gene of T. maritima. Our hypothesis is that TM1217 codes for a bifunctional protein that would act as a hydrogenase and a glutamate synthase.

Structure of the mbx operon in Pyrococcus species (a) and T. maritima (b). Genes are represented by arrows of length proportional to the gene length.
HGT of a Six-Gene Cluster to T. tengcongensis and D. gigas
In the same HBG000146 and HBG003659 families, another striking phylogenetic incongruency was detected. It involves T. tengcongensis, a hyperthermophilic bacterium, and D. gigas, a δ-proteobacterium. In both cases, these two bacteria are grouped together and the clade they form is grouped (bootstrap, >80%) with two methanogenic archaea: Methanosarcina mazei and M. barkeri (Fig. 1). Also, the two sequences are neighbors on the chromosome of T. tengcongensis and the analysis of their adjacent sequences allowed us to identify a cluster of six consecutive genes on the chromosome annotated as subunits of NADH:ubiquinone oxidoreductase (Table 2). In all trees built using these six genes, we found that T. tengcongensis and D. gigas are grouped with M. mazei and M. barkeri, this with a high bootstrap support (data not shown), again suggesting that the whole operon has been transferred. The order of these six genes is conserved in the four species considered. It has been experimentally demonstrated in M. barkeri (Künkel et al. 1998; Meuer et al. 1999; Meuer et al. 2002) and in D. gigas (Rodrigues et al. 2003) that they code for the Ech hydrogenase, an enzyme implied in energy metabolism. Therefore, as already mentioned by Rodrigues et al. (2003), the sequences of T. tengcongensis have been wrongly annotated as NADH:ubiquinone oxidoreductase, and the conservation of the synteny suggests that they encode a hydrogenase very similar to Ech. This has recently been confirmed by Soboh et al (2004), who have also demonstrated experimentally that the hydrogenase encoded was functional.
Discussion and Conclusion
Hydrogenases catalyze the reversible oxidation of molecular hydrogen and play a central role in microbial energy metabolism. They are mainly found in archaea and bacteria but a few are present in eukaryotes. On the basis of the transition-metal content, hydrogenases can be divided into three classes—[Fe]-hydrogenases, [NiFe]-hydrogenases, and metal-free hydrogenases—but the majority of them belong to the first two classes (Vignais et al. 2001).
Particularly, Mbh and Ech belong to a family of membrane-bound [NiFe]-hydrogenases that form a distinct group within the large family of [NiFe] hydrogenases (Vignais et al., 2001). This group includes also hydrogenases 3 and 4 from Escherichia coli, CO-induced hydrogenase from Rhodospirillum rubrum and Carboxydothermus hydrogenoformans, Eha and Ehb hydrogenase from Methanothermobacter species, and Ech hydrogenase from Methanosarcina barkeri (Hedderich 2004). These different hydrogenases are evolutionarily linked and recent reviews (Albracht and Hedderich 2000; Friedrich and Scheide 2000; Friedrich and Weiss 1997; Hedderich 2004) have shown that there is an evolutionary relationship between the membrane-bound [NiFe] hydrogenases and the energy conserving NADH:quinone oxidoreductase, also known as complex I. So it is not surprising that some of the families we have studied contain sequences belonging to different types of hydrogenases (e.g., sequences from the operons mbx and ech in the families HBG000146 and HBG003659).
Hydrogenases display highly modular structures and present a lot of duplications and rearrangements (Vignais et al. 2001). Therefore, as it is known that duplicated genes are more likely to be transferred (Hooper and Berg 2003), the existence of HGTs involving hydrogenases is not surprising. Moreover, many studies have reported HGTs involving such proteins, particularly transfers between archaea and bacteria (for review see Boucher et al. 2003).
The interesting fact here is that we have found HGTs events involving gene clusters corresponding to entire operons. Even if events like this have been described previously in different bacteria and archaea (Mhlanga-Mutangadura et al. 1998; Martin et al. 1998; Kennedy et al. 2001; Igarashi et al. 2001), the occurrence of independent transfers of related gene clusters to distinct genomes has not been previously described. Omelchenko et al. (2003) have demonstrated that the operon structures sometimes follow a mosaic evolution, by xenologous gene displacement or de novo assembly. That is not the case here: all cistrons share the same evolutionary history.
The mbx operon in the genome of P. furiosus contains genes coding for a putative membrane-bound [NiFe]-hydrogenase, but its activity has not been experimentally demonstrated. Interestingly, it is located at approximatively 5 kb from another operon, called mbh, which contains 14 genes and has a similar structure. In the case of mbh, its genes code for a membrane-bound [NiFe]-hydrogenase whose activity has been demonstrated (Sapra et al. 2000; Silva et al. 2000). This multiprotein complex, coupled with GAP:ferredoxin oxidoreductase, couples electron transfer to both proton reduction and proton translocation in P. furiosus (Sapra et al. 2003). Consequently, a possible origin for mbx may be a duplication of the mbh operon. As the transfer that occurred in T. maritima involved mbx, this could be indirect evidence of the fact that this operon codes for functional proteins in Pyrococcus species.
The function of the ech operon has been experimentaly charaterized in several organisms. In M. barkeri its genes code for a multisubunit membrane-bound [NiFe]-hydrogenase that plays a central role in the metabolism. The functions of this hydrogenase are multiple, including energy-conserving electron transport, as well as coupling of thermodynamically unfavorable reactions to reverse electron transport (Meuer et al. 2002). In D. gigas it has been shown that the hydrogenase it encodes could have a role in the process of energy conservation (Rodrigues et al. 2003). This operon is also present in another Desulfovibrio species: D. vulgaris Hildenborough. The corresponding hydrogenase could be involved in energy conservation functions and it may act as a hydrogen-producing enzyme in lactate metabolism with ferredoxin as redox partner. At last, the Ech hydrogenase of T. tengcongensis has recently been purified and characterized. It has been demonstrated that it favors hydrogen evolution over H2 uptake and it could participate in “proton respiration” (Soboh et al. 2004)
As T. tengcongensis and D. gigas belong to very distant taxonomic clades, the most parsimonious hypothesis to explain the fact they are clustered in our phylogenies is the occurrence of two independent horizontal transfers from a Methanosarcina-like organism to the genomes of the ancestors of these bacteria. A second scenario for the shared transfer in T. tengcongensis and D. gigas would be an initial transfer from an archaea to an ancestor of one of these bacterial lineages followed by a second bacterium to bacterium transfer. Also, it seems that the ech operon has been lost in some Desulfovibrio lineages since its transfer, as some species (such as D. desulfuricans G20) do not have the corresponding genes (Rodrigues et al. 2003). The fact that the transferred operons encode functional hydrogenases in T. tengcongensis and D. gigas is an indirect indication that the operon transferred in the genome of T. maritima could be functional too.
There are three possible interpretations for the branch lengths observed in the two clusters containing the hypothetical transfers: transfers may have occurred (i) from a species belonging to the Methanosarcina or to the Pyrococcus groups, respectively, and not yet sequenced or (ii) from an archaea belonging to another group of archaea not yet discovered, or (iii) it could be a transfer ancient enough that substantial evolution occurred in donor and acceptor since transfer.
References
SPJ Albracht R Hedderich (2000) ArticleTitleLearning from hydrogenases: location of a proton pump and of a second FMN in bovine NADH-ubiquinone oxidoreductase (Complex I) FEBS Lett 485 1–6 Occurrence Handle1:CAS:528:DC%2BD3cXot12qsL4%3D Occurrence Handle11086155
SF Altschul TL Madden AA Schaffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: a new generation of protein database search programs Nucleic Acids Res 25 3389–3402 Occurrence Handle10.1093/nar/25.17.3389 Occurrence Handle1:CAS:528:DyaK2sXlvFyhu7w%3D Occurrence Handle9254694
L Aravind RL Tatusov YI Wolf DR Walker EV Koonin (1998) ArticleTitleEvidence for massive gene exchange between archeal and bacterial hyperthermophiles Trends Genet 14 442–444 Occurrence Handle1:CAS:528:DyaK1cXns1ynur8%3D Occurrence Handle9825671
B Boeckmann A Bairoch R Apweiler MC Blatter A Estreicher E Gasteiger MJ Martin K Michoud C O’Donovan I Phan et al. (2003) ArticleTitleThe SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003 Nucleic Acids Res 31 365–370 Occurrence Handle10.1093/nar/gkg095 Occurrence Handle1:CAS:528:DC%2BD3sXhvFSmtL0%3D Occurrence Handle12520024
Y Boucher CJ Douady RT Papke DA Walsh ME Boudreau CL Nesbo RJ Case WF Doolittle (2003) ArticleTitleLateral gene transfer and the origins of prokaryotic groups Annu Rev Genet 37 283–328 Occurrence Handle1:CAS:528:DC%2BD2cXhsFGksA%3D%3D Occurrence Handle14616063
J Castresana (2000) ArticleTitleSelection of conserved blocks from multiple alignments for their use in phylogenetic analysis Mol Biol Evol 17 540–552 Occurrence Handle1:CAS:528:DC%2BD3cXisVSgt7g%3D Occurrence Handle10742046
V Daubin G Perrière (2003) ArticleTitleG+C3 structuring along the genome: a common feature in prokaryotes Mol Biol Evol 20 471–483 Occurrence Handle1:CAS:528:DC%2BD3sXkvFCnsrs%3D Occurrence Handle12654929
G Deckert PV Warren T Gaasterland WG Young AL Lenox DE Graham R Overbeek MA Snead M Keller M Aujay R Huber RA Feldman JM Short GJ Olsen RV Swanson (1998) ArticleTitleThe complete genome of the hyperthermophilic bacterium Aquifex aeolicus Nature 392 353–358 Occurrence Handle10.1038/32831 Occurrence Handle1:CAS:528:DyaK1cXit12qsL4%3D Occurrence Handle9537320
J Felsenstein (1989) ArticleTitlePHYLIP—Phylogeny inference package (Version 3.2) Cladistics 5 164–166
P Forterre (2002) ArticleTitleA hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein Trends Genet 18 236–237 Occurrence Handle1:CAS:528:DC%2BD38XjvFCgtr4%3D Occurrence Handle12047940
T Friedrich D Scheide (2000) ArticleTitleThe respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases FEBS Lett 479 1–5 Occurrence Handle10.1016/S0014-5793(00)01867-6 Occurrence Handle1:CAS:528:DC%2BD3cXlsFCit7w%3D Occurrence Handle10940377
T Friedrich H Weiss (1997) ArticleTitleModular evolution of the respiratory NADH:ubiquinone oxidoreductase and the origin of its modules J Theor Biol 187 529–540 Occurrence Handle1:CAS:528:DyaK2sXmtVGlsL4%3D Occurrence Handle9299297
J Gamieldien A Ptitsyn W Hide (2002) ArticleTitleEukaryotic genes in Mycobacterium tuberculosis could have a role in pathogenesis and immunomodulation Trends Genet 18 5–8 Occurrence Handle1:CAS:528:DC%2BD3MXpt1ahu7w%3D Occurrence Handle11750687
S Garcia-Vallvé A Romeu J Palau (2000) ArticleTitleHorizontal gene transfer in bacterial and archaeal complete genomes Genome Res 10 1719–1725 Occurrence Handle10.1101/gr.130000 Occurrence Handle11076857
O Gascuel (1997) ArticleTitleBIONJ: an improved version of the NJ algorithm based on a simple model of sequence data Mol Biol Evol 14 685–695 Occurrence Handle1:CAS:528:DyaK2sXksVahs7o%3D Occurrence Handle9254330
S Guindon O Gascuel (2003) ArticleTitleA simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood Syst Biol 52 696–704 Occurrence Handle10.1080/10635150390235520 Occurrence Handle14530136
S Guindon G Perrière (2001) ArticleTitleIntragenomic base content variation is a potential source of biases when searching for horizontally transferred genes Mol Biol Evol 18 1838–1840 Occurrence Handle1:CAS:528:DC%2BD3MXmt12hsLk%3D Occurrence Handle11504864
R Hedderich (2004) ArticleTitleEnergy-converting [NiFe] hydrogenases from archaea and extremophiles: ancestors of complex I J Bioenerg Biomembr 36 65–75 Occurrence Handle1:CAS:528:DC%2BD2cXitVOit7Y%3D Occurrence Handle15168611
DG Higgins JD Thompson TJ Gibson (1996) ArticleTitleUsing CLUSTAL for multiple sequence alignments Methods Enzymol 266 383–402 Occurrence Handle1:CAS:528:DyaK28Xltl2nt7w%3D Occurrence Handle8743695
SD Hooper O Berg (2003) ArticleTitleDuplication is more common among laterally transferred genes than among indigenous genes Genome Biol 4 R48 Occurrence Handle12914657
N Igarashi J Harada S Nagashima K Matsuura K Shimada KV Nagashima (2001) ArticleTitleHorizontal transfer of the photosynthesis gene cluster and operon rearrangement in purple bacteria J Mol Evol 52 333–341 Occurrence Handle1:CAS:528:DC%2BD3MXivVSisLo%3D Occurrence Handle11343129
R Jain MC Rivera JE Moore JA Lake (2003) ArticleTitleHorizontal gene transfer accelerates genome innovation and evolution Mol Biol Evol 20 1598–1602 Occurrence Handle1:CAS:528:DC%2BD3sXotlWjsrk%3D Occurrence Handle12777514
DT Jones WR Taylor JM Thornton (1992) ArticleTitleThe rapid generation of mutation data matrices from protein sequences Comput Appl Biosci 8 275–282 Occurrence Handle1:CAS:528:DyaK38Xlt1Okt7w%3D Occurrence Handle1633570
S Karlin AM Campbell J Mràzek (1998a) ArticleTitleComparative DNA analysis across diverse genomes Annu Rev Genet 32 182–225
S Karlin J Mràzek AM Campbell (1998b) ArticleTitleCodon usage in different classes of the Escherichia coli genome Mol Microbiol 29 1341–1355 Occurrence Handle1:CAS:528:DyaK1cXmsVKisLc%3D
SP Kennedy WV Ng SL Salzberg L Hood S DasSarma (2001) ArticleTitleUnderstanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence Genome Res 11 1641–1650 Occurrence Handle1:CAS:528:DC%2BD3MXnvFCmtrs%3D Occurrence Handle11591641
RJ Kinsella JO McInerney (2003) ArticleTitleEukaryotic genes in Mycobacterium tuberculosis? Possible alternative explanations Trends Genet 19 687–689 Occurrence Handle1:CAS:528:DC%2BD3sXpt1emsbo%3D Occurrence Handle14642748
LB Koski GB Golding (2001) ArticleTitleThe closest BLAST hit is often not the nearest neighbor J Mol Evol 52 540–542 Occurrence Handle1:CAS:528:DC%2BD3MXksFOiurs%3D Occurrence Handle11443357
LB Koski RA Morton GB Golding (2001) ArticleTitleCodon bias and base composition are poor indicators of horizontally transferred genes Mol Biol Evol 18 404–412 Occurrence Handle1:CAS:528:DC%2BD3MXhvVKrtrc%3D Occurrence Handle11230541
A Künkel JA Vorholt RK Thauer R Hedderich (1998) ArticleTitleAn Escherichia coli hydrogenase-3-type hydrogenase in methanogenic archaea Eur J Biochem 252 467–476 Occurrence Handle10.1046/j.1432-1327.1998.2520467.x Occurrence Handle9546662
NC Kyrpides GJ Olsen (1999) ArticleTitleArchaeal and bacterial hyperthermophiles: horizontal gene exchange or common ancestry? Trends Genet 15 298–299 Occurrence Handle1:CAS:528:DyaK1MXltVemsbs%3D Occurrence Handle10431189
ES Lander LM Linton B Birren et al. (2001) ArticleTitleInitial sequencing and analysis of the human genome Nature 409 860–921 Occurrence Handle10.1038/35057062 Occurrence Handle11237011
JG Lawrence H Ochman (1998) ArticleTitleMolecular archaeology of the Escherichia coli genome Proc Natl Acad Sci USA 95 9413–9417 Occurrence Handle1:CAS:528:DyaK1cXltlClsro%3D Occurrence Handle9689094
KS Makarova YI Wolf EV Koonin (2003) ArticleTitlePotential genomic determinants of hyperthermophily Trends Genet 19 172–176 Occurrence Handle1:CAS:528:DC%2BD3sXis12nsrY%3D Occurrence Handle12683966
K Martin G Morlin A Smith A Nordyke A Eisenstark M Golomb (1998) ArticleTitleThe tryptophanase gene cluster of Haemophilus influenzae type b: evidence for horizontal gene transfer J Bacteriol 180 107–118 Occurrence Handle1:CAS:528:DyaK1cXotVWj Occurrence Handle9422600
C Médigue T Rouxel P Vigier A Hénaut A Danchin (1991) ArticleTitleEvidence for horizontal gene transfer in Escherichia coli speciation J Mol Biol 222 851–856 Occurrence Handle1762151
J Meuer S Bartoschek J Koch A Kunkel R Hedderich (1999) ArticleTitlePurification and catalytic properties of Ech hydrogenase from Methanosarcina barkeri Eur J Biochem 265 325–335 Occurrence Handle1:CAS:528:DyaK1MXmsFKksLs%3D Occurrence Handle10491189
J Meuer HC Kuettner JK Zhang R Hedderich WW Metcalf (2002) ArticleTitleGenetic analysis of the archaeon Methanosarcina barkeri Fusaro reveals a central role for Ech hydrogenase and ferredoxin in methanogenesis and carbon fixation Proc Natl Acad Sci USA 99 5632–5637 Occurrence Handle1:CAS:528:DC%2BD38XjtFKltLk%3D Occurrence Handle11929975
T Mhlanga-Mutangadura G Morlin AL Smith A Eisenstark M Golomb (1998) ArticleTitleEvolution of the major pilus gene cluster of Haemophilus influenzae J Bacteriol 180 4693–4703 Occurrence Handle1:CAS:528:DyaK1cXlvVClsL8%3D Occurrence Handle9721313
KE Nelson RA Clayton SR Gill et al. (1999) ArticleTitleEvidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima Nature 399 323–329
CL Nesbo S L’Haridon KO Stetter WF Doolittle (2001) ArticleTitlePhylogenetic analyses of two “archaeal” genes in Thermotoga maritima reveal multiple transfers between archaea and bacteria Mol Biol Evol 18 362–375 Occurrence Handle1:CAS:528:DC%2BD3MXhvVKrtrs%3D Occurrence Handle11230537
CL Nesbo KE Nelson WF Doolittle (2002) ArticleTitleSuppressive subtractive hybridization detects extensive genomic diversity in Thermotoga maritima J Bacteriol 184 4475–4488 Occurrence Handle1:CAS:528:DC%2BD38XlvV2gs78%3D Occurrence Handle12142418
P Nicolas L Bize F Muri M Hoebeke F Rodolphe SD Ehrlich B Prum P Bessières (2002) ArticleTitleMining Bacillus subtilis chromosome heterogeneities using hidden Markov models Nucleic Acids Res 30 1418–1426 Occurrence Handle1:CAS:528:DC%2BD38XisVakt70%3D Occurrence Handle11884641
MV Omelchenko KS Makarova YI Wolf IB Rogozin EV Koonin (2003) ArticleTitleEvolution of mosaic operons by horizontal gene transfer and gene displacement in situ Genome Biol 4 R55 Occurrence Handle12952534
G Perrière L Duret M Gouy (2000) ArticleTitleHOBACGEN: database system for comparative genomics in bacteria Genome Res 10 379–385 Occurrence Handle10720578
R Rodrigues FM Valente IA Pereira S Oliveira C Rodrigues-Pousada (2003) ArticleTitleA novel membrane-bound Ech [NiFe] hydrogenase in Desulfovibrio gigas Biochem Biophys Res Commun 306 366–375 Occurrence Handle1:CAS:528:DC%2BD3sXksVyksbc%3D Occurrence Handle12804572
R Sapra MF Verhagen MW Adams (2000) ArticleTitlePurification and characterization of a membrane-bound hydrogenase from the hyperthermophilic archaeon Pyrococcus furiosus J Bacteriol 182 3423–3428 Occurrence Handle10.1128/JB.182.12.3423-3428.2000 Occurrence Handle1:CAS:528:DC%2BD3cXjvFWqurk%3D Occurrence Handle10852873
R Sapra K Bagramyan MW Adams (2003) ArticleTitleA simple energy-conserving system: proton reduction coupled to proton translocation Proc Natl Acad Sci USA 100 7545–7550
PJ Silva EC Ban Particlevan den H Wassink H Haaker B Castro Particlede FT Robb WR Hagen (2000) ArticleTitleEnzyme of hydrogen metabolism in Pyrococcus furiosus Eur J Biochem 267 6541–6551 Occurrence Handle10.1046/j.1432-1327.2000.01745.x Occurrence Handle1:CAS:528:DC%2BD3cXotF2qsbY%3D Occurrence Handle11054105
B Soboh D Linder R Hedderich (2004) ArticleTitleA multisubunit membrane-bound [NiFe] hydrogenase and a NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis Microbiology 150 2451–2463 Occurrence Handle1:CAS:528:DC%2BD2cXmtFGru7s%3D Occurrence Handle15256587
MJ Stanhope A Lupas MJ Italia KK Koretke C Volker JR Brown (2001) ArticleTitlePhylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates Nature 411 940–944 Occurrence Handle1:CAS:528:DC%2BD3MXkvVWhurk%3D Occurrence Handle11418856
K Strimmer A Haeseler Particlevon (1996) ArticleTitleQuartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies Mol Biol Evol 13 964–969 Occurrence Handle1:CAS:528:DyaK28XltlSmsLk%3D
PM Vignais B Billoud J Meyer (2001) ArticleTitleClassification and phylogeny of hydrogenases FEMS Microbiol Rev 25 455–501 Occurrence Handle1:CAS:528:DC%2BD3MXmt1Kls7Y%3D Occurrence Handle11524134
B Wang (2001) ArticleTitleLimitations of compositional approaches to identifying horizontally transferred genes J Mol Evol 53 244–250 Occurrence Handle1:CAS:528:DC%2BD3MXnt1Ghs7k%3D Occurrence Handle11523011
Acknowledgments
This work has been supported by CNRS and MENRT. A.C. is the recipient of a fellowship from MENRT. All phylogenetic reconstructions were done at the IN2P3 computational center (Villeurbanne, France).
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Siv Andersson
Rights and permissions
About this article
Cite this article
Calteau, A., Gouy, M. & Perrière, G. Horizontal Transfer of Two Operons Coding for Hydrogenases Between Bacteria and Archaea. J Mol Evol 60, 557–565 (2005). https://doi.org/10.1007/s00239-004-0094-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0094-8