
Abstract
We isolated the intracellular parasitic bacterium Caedibacter taeniospiralis from cultures of the freshwater ciliate Paramecium tetraurelia strain 298. Plasmid pKAP298 as well as the total RNA were isolated from the bacteria. pKAP298 was totally sequenced (49.1 kb; NCBI accession number AY422720). From southern blots of pKAP-fragments and Digoxigenin-labeled cDNA of the Caedibacter-RNA, we generated transcription maps of pKAP298. The observed transcription activity indicated functions of the plasmid besides the synthesis of the R-body, a complex protein inclusion associated with toxic effects of Caedibacter cells on host paramecia. We identified 63 potential protein coding regions on pKAP298, and a novel transposon as well as known transposons were characterized. A group II intron was identified. Homologies with putative phage genes were detected on pKAP298 that direct to the evolution of pKAP298 from a bacteriophage. This original phage most probably belonged to the Caudovirales. Hints on a toxin coding region of pKAP298 are given: a protein with homology to the Soj-/ParA-family also has homologies to a membrane associated ATPase, which is involved in eukaryotic ATPase dependent ion carriers and may be associated with toxic effects on paramecia ingesting this protein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A wide diversity of intracellular bacteria live in protozoa (Preer and Preer 1964; Görtz 1996; Görtz and Brigge 1998). Some of the bacteria are closely related to pathogens and exploit the host cell functions just as invasive agents in eukaryotes (Winkler and Neuhaus 1999; Linka et al. 2003). Few of these intracellular bacteria can be grown in vitro, and therefore investigations with traditional microbiological methods are not possible.
Intracellular bacteria of the genus Caedibacter are characterized by their ability to produce R-bodies (refractile bodies), their ability to produce a toxin, and their obligate endobiotic nature (Pond et al. 1989). Caedibacter taeniospiralis (γ-Proteobacteria; Thiotrichales [Beier et al. 2002]) inhabits the freshwater ciliate Paramecium tetraurelia as a host. R-Body-containing Caedibacter cells are toxic to paramecia that do not contain endobionts or not the same species of Caedibacter bacteria. Therefore, while limiting the reproduction of their host, the endobionts also give competitive advantages to Paramecium under some circumstances (Landis 1981, 1987; Kusch et al. 2002). Caedibacter species are infected by phages or plasmids, which are responsible for the production of R-bodies and probably the toxin. Five species of the genus are described (Preer et al. 1974; Quackenbush 1978; Schmidt et al. 1987a).
DNA hybridizations (Quackenbush 1977, 1978) and 16S rRNA sequences (Beier et al. 2002) showed little similarity among different Caedibacter species, indicating that the genus Caedibacter may be polyphyletic, with, e.g., C. caryophilus belonging to the α-Proteobacteria. Nevertheless, extrachromosomal elements coding for R-bodies in different Caedibacter species seem to be more closely related than their hosts (Quackenbush et al. 1986a). R-Body production occurs in nonendobiotic bacteria also, including pseudomonads (Pond et al. 1989). Most probably different bacterial genera gained (or were infected by) equal or similar extrachromosomal elements during their evolution.
Among species of the genus Caedibacter, C. taeniospiralis is the only one owning a plasmid (pKAP) as an R-body coding extrachromosomal element (Dilts 1976; Quackenbush et al. 1983a,b). Plasmid pKAP is present in all strains of Caedibacter taeniospiralis, but varying sizes of pKAP (41.5 to 50.5 kb) occur. The plasmids of C. taeniospiralis strains differ only by the insertion of transposons 1.5 and/or 7.5 kb in size at different plasmid locations (Quackenbush et al. 1986b). Tn4501 and Tn4501/2 are of 1.5 kb and very similar. Some pKAP plasmids additionally contain Tn4502, which also is very similar to Tn4501. It is not present in pKAP298. Tn4503 is of 7.5 kB and owns a region with homology to the smaller transposons. Presumably Tn4503 is a 6-kB transposon with Tn4501/2 inserted. The transposons are present in the genome of Caedibacter taeniospiralis also. From these transposons, only a gene for a transposase has been characterized so far. On pKAP116 and pKAP47 the R-body coding genes were identified (Heruth et al. 1994); other genes and the origin of pKAP plasmids were unknown.
The pKAP plasmids probably evolved from bacteriophages. R-Bodies from Caedibacter species except C. taeniospiralis are associated with phages or capsomer-like structures (Preer et al. 1972, 1974; Pond et al. 1989; Kusch et al. 2000). Bacteriophages apparently direct the synthesis of R-bodies and toxin, as does pKAP in C. taeniospiralis. They are evidently noninfective, and no evidence exists that bacterial lysis occurs (Pond et al. 1989), with the exception of Caedibacter caryophilus in the host Paramecium caudatum (Schmidt et al. 1987b). The R-body coding region is of 1.8 kb and consists of the three genes rebA, rebB, and rebC. They code for proteins of 18, 13, and 10 kDa. A putative fourth gene, rebD, may be involved in R-body production and expression of the toxin (Heruth et al. 1994). To learn about the evolutionary origin of R-body coding plasmids and about functions of pKAP plasmids besides R-body production, we isolated and characterized the plasmid pKAP298 of C. taeniospiralis strain 298. Total sequence and transcription activity were analyzed to identify potential gene products, including a toxin coding region. Transposon regions were characterized to evaluate the evolutionary relationship of different pKAP plasmids.
Methods
Cultivation and Isolation of Caedibacter taeniospiralis
The host of Caedibacter taeniospiralis, Paramecium tetraurelia strain 298, was raised in a hay medium (10 mM NaH2PO4, 10 mM Na2HP04, 10% [v/v] hay concentrate). The hay concentrate was made by autoclaving 100 g of hay in 1 L of destilled water for 1 h, followed by filtration through gauze. The medium was inoculated with Klebsiella minuta and incubated for 1 day at 37°C, before adding P. tetraurelia. Every other day, the culture volume was doubled and the cultures were filtered through gauze. When the final volume was reached, the cultures were incubated for a further 10 days at 14°C. The presence of C. taeniospiralis was controlled by microscopy and PCR detection according to Kusch et al. (2002).
For the isolation of C. taeniospiralis from host cells we reduced the volume of P. tetraurelia cultures (typically 32 L) to 400 ml by centrifugation in a cream separator (Klärseparator KA 05-00-105; Westfalia Separator, Germany). The Paramecia were pelleted by 5 min of centrifugation at 3000g in an oil test centrifuge (HNS II; Damon IEC Division). Caedibacter taeniospiralis were isolated from Paramecium tetraurelia by an adaptation of the procedures described by Mueller (1963). Paramecium pellets were homogenized and taken up in buffer A (150 mM sorbitol, 10 mM KH2PO4, 10 mM K2HPO4). The homogenate was centrifuged (4500g, 5 min, 4°C) and washed four times in buffer A. The homogenate was mixed with 1 g Ecteola 23 (Serva) for 2 min on ice and subsequently chromatographed on a column with buffer A. The adsorbent Ecteola 23 was equilibrated twice for 30 min in 100 ml buffer A before use. The flow-through containing the Caedibacter bacteria was pelleted by centrifugation (4500g, 5 min, 4°C).
Isolation and Modification of DNA and RNA from Caedibacter taeniospiralis
Plasmid Isolation
The plasmid pKAP298 was isolated from C. taeniospiralis using the Plasmid Midi Kit (Quiagen), according to the protocol supplied with the kit. If Southern blots were to be made up, the plasmid isolate was not purified through the column delivered with the kit (to prevent possible loss of DNA), but by Phenol/chloroform extraction (equal volumes of phenol/chloroform; two washes in an equal volume of chloroform) followed by precipitation with isopropanol (equal volume of isopropanol; 1 hr at –20°C; centrifugation at 12,000g for 15 min; two washes in 70% ethanol).
Isolation of RNA
The total RNA from C. taeniospiralis was isolated with pegGOLD RNAPure reagent (Peqlab), according to the manual. To remove contaminating DNA (e.g., plasmid pKAP298), each isolate was digested with DNAse I (RNAse free; 10 U DNAse I/μg RNA; 30 min at 37°C). The DNAse I was heat-inactivated thereafter (5 min at 95°C).
Transcription of Total RNA in cDNA
One microgram of each RNA isolate was transcribed in cDNA using the RevertAid H Minus First Strand cDNA Synthesis kit (MBI Fermentas), according to the manual.
Digoxigenin Labeling of cDNA
All cDNA-labeling reactions and hybridizations were performed with the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche). After denaturation for 10 min at 99°C, 1μg of cDNA and the labeling reagent were mixed and incubated for 20 h at 37°C.
Subcloning and Amplification of pKAP298 Fragments in E. coli
pKAP298 isolates were digested with restriction endonucleases and separated on agarose gels. The use of suitable restriction enzymes according to restriction maps of pKAP298 established by Quakenbush (1983) assured the generation of fragments large enough for isolation and cloning, without the danger of loss of very small fragments. DNA fragments were isolated from the gels and purified with the NucleoSpin Extract 2-in-1 kit (Machery & Nagel). Purified fragments to be cloned were ligated in plasmid vector pHSG298 (Takara). If a fragment had identical ends, the vector pHSG298 was dephosphorylated before ligation. Recombinant vectors were transformed into E. coli by electroporation and those recombinants that carried the desired inserts were identified by restriction digests. Positive recombinants were grown in LB medium (40 ml each) to their maximum density and subsequently the recombinant vectors were isolated and purified using the Plasmid Midi Kit (Quiagen).
Southern Hybridizations
Whole pKAP298 plasmid DNA or pKAP298 fragments amplified in E. coli were digested with appropriate restriction endonucleases and separated on an 1.4% agarose gel using 1 × TAE (40 mM Tris/HCl, 20 mM glacial acid, 5 mM EDTA, pH 7.5) as running buffer at 3.5 V/cm. The gels were first stained with ethidium bromide and treated for 20 min each in solutions A (250 mM HCl), B (500 mM NaOH, 3 M NaCl), and C (500 mM Tris-HCl, 1.5 M NaCl, pH 7). Gels were blotted overnight to nylon membranes (Hybond N; Amersham) in 10x SSC. DNA was fixed by UV exposure at a dose of 200 mJ/cm2. Prehybridizations were performed at 39°C for 30 min. Hybridizations took place for 24 h at 39°C, followed by two stringency washes (20 min in 2x SSC at RT and 20 min in 0.5x SSC at 65°C). Detections were performed according to the kit manual and documented on BioMAX Light films (Kodak). The films were analyzed and pKAP298 fragments with transcriptional activity were identified.
DNA Analysis
Sequencing was performed four times on an ABI 377 Sequencer (Applied Biosystems) by single-strand primer-walking with an error rate of 0.1%. To assure assembling of sequenced fragments in the right order and orientation, we generated virtual restriction maps from the obtained sequences. These maps were compared to a restriction map of pKAP298 established by Quakenbush (1983). The location of transposons also was compared to previous results (Quackenbush et al. 1986b; Pond et al. 1989). The pKAP298 sequence was searched for putatively coding open reading frames (ORF) using the GeneMark.hmm program (Lukashin and Borodovski 1998). Identified ORFs were searched for homologies (bases and aminoacids) to already known sequences with the BLAST database (NCBI). G/C content was calculated with GeeCee (EMBOSS package). Transposons were identified by searching for inverted repeats (with Einvert; EMBOSS package) and putative transposases located within the sequence between the inverted repeats.
Results
Since Caedibacter species so far cannot be grown outside their host paramecia, we isolated C. taeniospiralis from an approximately 3000-L culture volume of P. tetraurelia (1200–1500 cells/ml). From isolated C. taeniospiralis we purified the plasmid pKAP298 (0.26–0.48 μg/L of paramecium culture; depending on the purification method, either plasmid Midi Kit or phenol/chloroform extraction; Fig. 1A) as well as the total RNA (Fig. 1B). pKAP298 was cleaved by various combinations of several restriction enzymes (AvaI, BamHI, EcoRI, EcoRV, HindIII, KpnI, PstI, SacI, SalI, XhoI) and resulting fragments were subcloned in E. coli. From fragments of pKAP298 (directly cleaved or subcloned fragments), Southern blots were prepared (Figs. 1C and E).

Isolation of plasmid pKAP298 (A; cleaved with the restriction enzyme PstI) and of total RNA (B) from Caedibacter taeniospiralis. An example of a southern blot of subcloned pKAP fragments (C) and hybridization with labeled cDNA (D) is given. E Southern blot of a pKAP298 isolate cleaved with either EcoRI + SacI (left) or EcoRI + XhoI (right). F Corresponding hybridization with labeled cDNA. M1, λ19-mix marker (lowest band = 8271 bp); M2, 100-bp ladder-plus marker (uppermost = 3000 bp); M3, EcoT13 marker (uppermost = 19,329 bp).
Transcription Analysis
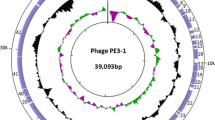
To analyze the transcription activity and to identify potential gene products, we performed hybridization experiments with the Southern blots of pKAP298 fragments. Therefore, the isolated total RNA from C. taeniospiralis after digestion of contaminating DNA by DNAseI was transcribed into cDNA. The obtained cDNA was labeled with digoxigenin dUTP and used for 150 hybridizations with different pKAP298 Southern blots (Figs. 1D and F). From the detection of cDNA bound to pKAP298 fragments we established a transcription map of pKAP298. After sequence analysis of pKAP298 we identified the potential genes which were transcriptionally active (Fig. 2).

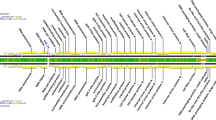
Map of transcription activity, ORFs, and transposons of the total plasmid pKAP298 (49112 bp). The map starts with the only BamHI restriction site, defined as bp 0. Each of the five double segments gives 10 kbp, except the last one, with 9.112 kbp. The upper part of each double segment shows regions with detected transcription activity above the line and transposons below the line. Base pair (Bp) numbers belong to vertical lines below the indicated Bp values. Some restriction sites are also given (below Bp numbers). The lower parts of each double segments show the detected ORFs, with the direction of transcription indicated by arrows at the left. Putative functions are given for some ORFs.
Sequence Analysis
Plasmid pKAP298 was totally sequenced in part from fragments subcloned in E. coli, as well as from directly isolated plasmid from Caedibacter taeniospiralis. The sequence was submitted to GenBank (NCBI; accession number AY422720). The plasmid is of 49,112 base pairs and contains 37% G+C. A restriction map with exact positions of cleavage sites was generated (Table 1). Using this map we were able to place restriction fragments with detected transcription activity to exact sequences of the Caedibacter plasmid (Fig. 2). Comparisons of the total pKAP298 sequence to other whole plasmids or phages gave no matches. Further sequence analyses revealed 63 potentially coding regions of pKAP298 (Tables 2 and 3). Of these 63 ORFs, 56 begin with the base triplett ATG as a start codon, 4 begin with GTG, and 3 with a TTG. Of the recognized ORFs, 23 have significant sequence homologies to known genes from Caedibacter species or other organisms (Table 2).
ORFs 1–3 and 63 code for proteins RebA, RebB, RebC, and RebD, which constitute the R-body of Caedibacter taeniospiralis (Heruth et al. 1994). RebA of pKAP298 differs from RebA coded by pKAP116 or pKAP47 in only one amino acid; RebD differs in six amino acids. ORF 53 is a transposase of transposon Tn4501. All other potentially coding regions of pKAP298 were unknown so far for pKAP plasmids. ORF 7 may be an incomplete putative transposase/recombinase that originally was involved in the catalysis of transposition of transposon Tn4503. ORF 8 also is a putative transposase, which may be involved in transposition of Tn4503. ORFs 9–11 together constitute a transposase that is similar to the transposase from transposon Tn4501. The three ORFs on pKAP298 are located within transposon Tn4501/2, which itself lies on Tn4503 (Fig. 2). ORF12 is located in transposon Tn4503 and may be a site-specific recombinase/resolvase that directs to a replicative transposition of Tn4503. ORF24 has homologies to a putative bacteriophage portal protein that may cooperate in the transfer of the phage genome to a phage capsid. ORF 25, as a putative DNA packaging protein, may also contribute to the transfer of the phage genome to a capsid. ORF 32 is a putative transposase of transposon Tn4504. ORF 39 has homologies to DNA binding proteins and an integration host factor. ORF 42 may be a phage-related minor tail protein of unknown function. ORF 43 is similar to Soj/ParA proteins and may be involved in the replication/segregation of pKAP298. ORF 44 has homologies to GerE bacterial regulatory proteins of the luxR family. ORF 47 may be a reverse transcriptase gene. It may represent a part of a group II intron. ORF 48 is located on pKAP298, directly neighboring the putative reverse transcriptase (ORF 47), and may also belong to the group II intron. ORFs 49, 50, and 54 have homologies to exodeoxyribonuclease VIII (RecE) of a prophage (CP-933R) and of E. coli. ORFs 49, 50, and 54 most probably belonged to one coding sequence, which now is interrupted by the insertion of transposon Tn4501, as well as ORFs 51 and 52, whose functions are still unknown.
We identified three known and one so far unknown (Tn4504) transposons on pKAP298. Tn4501 is located between base 42,862 and base 44,301 (=1439 bp), with ORF 53 as the putative transposase. Tn4501/2 is very similar to Tn4501 and lies between position 8756 and position 10,196 (=1440 bp), with ORFs 9–11 possibly being the original but now interrupted transposase. Tn4503 is between base 5591 and base 13,333 (=7742 bp). ORF 8 possibly is its transposase. This sequence, as well as ORFs 12–14, lies on Tn4503. The putative transposon Tn4504 is located between position 25,381 and position 33,379 (=7998 bp). The length of the inverted repeat sequence directs to a nonreplicative composed transposon of type Tn5. ORF 32 may be the putative transposase sequence. ORFs 26–40 are located on Tn4504.
Discussion
Transcription Activity of pKAP298
The plasmid pKAP298 of the intracellular parasite Caedibacter taeniospiralis was characterized to be more active in transcription as known so far. Until recently only the R-body coding sequences were known to code for proteins (Heruth et al. 1994). Stress conditions induce the transcription of these genes (Pond et al. 1989) that lie around the only BamHI restriction site present in the plasmid (Heruth et al. 1994). The observed further transcription activity of pKAP298 indicates functions besides R-body synthesis. Regions active in transcription contained genes with homology to several phage proteins. This fact directs to the synthesis of phages or incomplete phages in Caedibacter taeniospiralis also. ORF 42 has homology to a phage minor tail protein and may code for helical structures associated to R-bodies in some Caedibacter species (Preer et al 1974). ORF 43 has homology to a Soj/ParA enzyme and may therefore contribute to the replication/segregation of pKAP298 (Davey et al. 1994). ParA and ParB homologues were also found in plasmid prophage pKO2 of Klebsiella oxytoca (Casjens et al. 2004) and in E. coli plasmid prophage P1 (Abeles et al. 1985). Possible regulatory sequences like the detected ORFs 39 and 44, with homology to integration host factors or GerE proteins (Kirndorfer et al. 1998; Zheng et al. 1992), may be responsible for the repression or activation of transcription of coding regions.
Evolutionary Origin of pKAP298
We did not detect a significant homology of the total pKAP298 to other plasmids, phages, or genomes, indicating a so far unique molecular structure of pKAP298. Yet the occurrence of sequences with homology to phage proteins directs to the derivation of pKAP298 from a former phage, as hypothesized by Preer et al. (1974). Prophages of some temperate phages, e.g., of coliphages P1 (Ikeda and Tomizawa 1968; Walker and Walker 1975) and N15 (Ravin and Shulga 1970), also are autonomous plasmids. Furthermore, Casjens et al. (2004) reported that the Klebsiella oxytoca linear plasmid pKO2 is a prophage. In pKAP298, besides the transcribed ORFs with phage homology mentioned above, ORF 25 has homology to a putative DNA packaging protein GP2 (Galibert et al. 2001) and similar proteins as well as terminases of other phages (Mmolawa et al. 2003; Vander Byl and Kropinski 2000), which are involved in the transport of viral genomes into phage capsids (Fujisawa et al. 1997). A putative phage portal protein is present on pKAP298 also (ORF 24). Phage portal proteins are known to be part of a connector between head and tail of phages and are involved in the transport of viral genomes into phage capsids too (Black 1989). ORFs 49, 50, and 54 all have homology to a RecE protein of prophage VT1-Sakai from a strain of E. coli (Yokoyama et al. 2000) and of lambdoid phage HB-4 (Kim et al. 1999). The three sequences presumably were one sequence or lay together before the insertion of transposon Tn4501 (Quackenbush 1983; Quackenbush et al. 1986a,b). The similar RecE protein from diverse phages is important for their recombination (Shiraishi et al. 2002; Zhang et al. 2003). If the putative RecE protein of pKAP298 was involved in the integration of the former phage into the host genome, e.g., via induction of breakage of the DNA double helix (Zhang et al. 2003), then the phage should have lost this ability.
ORF 47 has a high homology to the protein RetA from a broad–host range plasmid (Kulaeva et al. 1998) that was characterized as a group II intron (Dai et al. 2002). Homology exists to other group II intron proteins (maturase) also (Nelson et al. 2002), similarly as for ORF 48 of pKAP298. By reverse transcriptase or maturase the group II intron RNA becomes transcribed into DNA and often is integrated at a distinct site of the genome (Klein et al. 2002; Lehmann and Schmidt 2003). We observed homology to phage integrases for ORF 12 (Read et al. 2002), which are active in the integration of phage genomes into host genomes. Yet since ORF 12 is located on transposon Tn4503, it presumably originates from the Caedibacter taeniospiralis genome. It also has homology to a site-specific recombinase from transposon Tn5401 of Bacillus thuringiensis (Baum 1994). Other sequences also may stem from transposition events, except ORFs 24, 25, 42, 49, 50, and 54, with homology to phage sequences. We did not detect sequences with homology to proteins that are involved in the phage-induced lysis of a host cell. Schmidt et al. (1987b) presumed from the occurrence of R-bodies in the macronucleus of C. caryophilus-infected Paramecium caudatum that bacteriophages of C caryophilus induced host cell lysis. In C. taeniospiralis induced cell lysis was never observed.
All classified bacteriophages that have sequence homology to pKAP298 (bacteriophage 933W, enterobacteriophage P22, mycobacteriophage Che9c, Pseudomonas aeroginosa phage PaP3, Salmonalla typhimurium bacteriophage ST64T) are from the order Caudovirales. Common features of the Caudovirales are tails and linear double-stranded DNA (Maniloff and Ackermann 1998). The genome size of pKAP298 without transposons (38.5 kb) is most similar to that of the Podoviridae among the different Caudovirales families. Yet it is still unknown whether the putative pKAP298 phage lost parts of its genome or has integrated sequences.
Transposons Tn4501, Tn4501/2, and Tn4503 that we identified on pKAP298 are homologous to transposons of pKAP plasmids of Caedibacter taeniospiralis strains and probably stem from the host’s genome. This was evident from observations of spontaneous insertions of those transposons into pKAP plasmids and from hybridizations of the transposons and genomic DNA (Dilts and Quackenbush 1986; Quackenbush et al. 1986b). Our sequence data showed that Tn4501/2 is very similar to Tn4501, as supposed by Quackenbush et al. (1986a, b). The detected putative transposase (ORFs 9–11) is fragmented and therefore Tn4501/2 may not be able for a transposition. The newly discovered putative transposon Tn4504 as well as a group II intron seems not to originate from C. taeniospiralis. Regions of similar restriction sites have been described for all pKAP plasmids known so far, but spontaneous insertions were never observed (Quackenbush 1983).
The plasmid pKAP298 may have evolved from a bacteriophage to a broad–host range plasmid, since some host strains classified as C. taeniospiralis have DNA homologies of 41% only, whereas in others it is 80% (Quackenbush 1977, 1978). Some of the ORFs we identified to originate from a phage have homologies to proteins of the order Rhizobiales (Kaneko et al. 2002; Galibert et al. 2001), to which the original host may have belonged.
Apparently pKAP298 and its R-body genes are related to a bacteriophage that does not have a complete set of phage head and tail genes. In several other bacteria, sequence analysis and electron microscopy revealed a common ancestry for some bacteriocins, bacterial products with specific bactericidal activity, and bacteriophages (e.g., Strauch et al. 2001; Jabrane et al. 2002). These bacteriocins have been regarded as defective bacteriophages, which might have arisen from temperate phages by several successive mutations (Bradley 1967; Daw and Falkiner 1996). Data on the gene organization of bacteriocins from Pseudomonas aeroginosa suggested that they are not simple defective phages but are phage tails that have been evolutionarily specialized as bacteriocins (Nakayama et al. 2000). Different bacteria obviously use phage structural genes for a bacteriocin production, similar to the case of the Caedibacter-derived R-body that has Paramecium toxicity. Therefore, loss of phage structural genes in pKAP298 seems not to result from random processes that finally prevented phage production. Instead the present genes should come from adaptive selection of a toxic function that preserved this genetic element.
Another example where bacterial functions may have evolved from phage genes is the gene transfer agent (GTA) of Rhodobacter capsulatus. GTA is a bacteriophage-like particle that transfers random 4.5-kb fragments of host genomic DNA between R. capsulatus cells (Marrs 1974; Yen et al. 1979). Several GTAs that resemble transducing phages have been found in diverse prokaryotes. None of these agents have lytic activity. Production of GTA results from expression of genes that are similar to phage genes, yet tanscription of these genes is dependent on two cellular signaling proteins in a growth phase-dependent manner. The genes that encode the proteins that make up the GTA particle are located on the R. capsulatus chromosome. These genes map to a cluster of approximately 15 kb that contains 15 ORFs. The sequence similarities of the GTA ORFs to known phage genes indicate that the GTA structural gene cluster encodes the proteins that make up the head and tail structure of GTA particles. Therefore, GTA could be a cellular genetic exchange vector that evolved from a phage. An alternative possibility is that the GTA genes represent an evolutionary precursor of a genuine phage genome. The control of the production of GTA by cellular proteins is consistent with the hypothesis that GTA is a cell-evolved entity (Lang and Beatty 2001). Sequences of homologues of GTA structural gene cluster and GTA regulatory genes from different α-proteobacteria revealed that structural and regulatory genes were present in a single progenitor bacterium. Evolutionary relationships of GTA structural proteins to (pro)phage proteins suggested a predominantly vertical descent of GTA-like sequences and little gene exchange with (pro)phages (Lang et al 2002). The presence of genes for R-body production in very diverse bacteria hints at their evolution from phages. R-Body production in Caedibacter species also occurs predominantly in a stationary phase. Analysis of the Caedibacter genome, e.g., for genes for regulatory proteins, could shed light on the question whether the evolution of pKAP298 involved gene transfer between the host and a phage genome.
A Putative Toxin-Coding Region
Associated with a stress induced R-body production in Caedibacter species is a probably pKAP-coded subsequent toxicity to paramecia ingesting R-bodies (Pond et al. 1989; Dilts and Quackenbusch 1986). Gibson (unpublished; in Pond et al. 1989), using protein chromatography, observed the toxin in cultures of P. tetraurelia 51 to be a protein of 20–25 kDa. Transcribed regions of pKAP298 with ORFs of that size are ORFs 7 (22.4 kDa), 9 (20.3 kDa), and 43 (24.6 kDa). Since ORFs 7 and 9 are on transposons Tn4503 and Tn4501/2, and C. taeniospiralis strains with plasmids that do not contain this transposon are toxic also, these ORFs cannot code for the toxin. ORF 43 codes for a protein with homology to the Soj-/ParA family. As an ATPase it may function in the distribution of pKAP298 on daughter cells after host divisions (Motallebi-Vesharesh 1990) and may interfere with the distribution of the host genome due to misrelations of ParA and ParB proteins (Easter et al. 2002), leading to the observed low reproduction rates observed for Caedibacter species (Preer et al. 1972). The toxic effect of the ORF 43 product on paramecia ingesting this protein might be associated with homologies to a membrane-associated ATPase, which is involved in eukaryotic ATPase-dependent ion carriers (Zhou et al. 2000). The toxin presumably has effects on the Paramecium cell membrane, where it might disturb the osmoregulative abilities of the ciliate (Butzel et al. 1962) and leads to hump-killing or vacuolization as a typical killing mechanism (Preer et al. 1974). While the ORF 43 codes for a protein that might have features enabling it to function as a toxin in eukaryotic host cells, we cannot exclude that other pKAP sequences code for the toxin, and further studies are necessary concerning this question.
References
AL Abeles SA Friedman SJ Austin (1985) ArticleTitlePartition of unit-copy miniplasmids to daughter cells. III. The DNA sequence and functional organization of the P1 partition region J Mol Bio 185 261–272
JA Baum (1994) ArticleTitleTn5401, a new class II transposable element from Bacillus thuringiensis J Bacteriol 176 2835–2845
CL Beier M Horn R Michel M Schweikert H-D Görtz M Wagner (2002) ArticleTitleThe genus Caedibacter comprises endobionts of Paramecium spp. related to the Rickettsiales (Alphaproteobacteria) and to Francisella tularensis (Gammaproteobacteria) Appl Environ Microbiol 68 6043–6050
LW Black (1989) ArticleTitleDNA packaging in dsDNA Bakteriophages Annu Rev Microbiol 43 267–292
DE Bradley (1967) ArticleTitleUltrastructure of bacteriophage and bacteriocins Bacteriol Rev 31 230–314
HM Butzel A Pagliara (1962) ArticleTitleThe effect of biochemicals inhibitors upon the killer sensitive system in Paramecium aurelia Exp Cell Res 27 382–395
SR Casjens EB Gilcrease WM Huang KL Bunny ML Pedulla ME Ford JM Houtz GF Hatfull RW Hendrix (2004) ArticleTitleThe pKO2 linear plasmid prophage of Klebsiella oxytoca J Bacteriol 186 1818–1832
L Dai S Zimmerly (2002) ArticleTitleCompilation and analysis of group II intron insertions in bacterial genomes: evidence for retroelement behavior Nucleic Acids Res 30 1091–1102 Occurrence Handle10.1093/nar/30.5.1091 Occurrence Handle1:CAS:528:DC%2BD38XitVOisb8%3D Occurrence Handle11861899
MJ Davey BE Funnell (1994) ArticleTitleThe P1 plasmid partition protein ParA. A role for ATP in site-specific DNA binding J Biol Chem 269 29908–29913
MA Daw FR Falkiner (1996) ArticleTitleBacteriocins: nature, function and structure Micron 27 467–479
JA Dilts (1976) ArticleTitleCovalently closed, circular DNA in kappa endobionts of Paramecium Genet Res 27 161–170
JA Dilts RL Quackenbush (1986) ArticleTitleA mutation in the R-body-coding sequences destroys expression of the killer trait in P. tetraurelia Science 232 641–643
J Easter JW Gober (2002) ArticleTitleParB-stimulated nucleotide exchange regulates a switch in functionally distinct ParA acitvities Mol Cell 10 427–434
H Fujisawa M Morita (1997) ArticleTitlePhage DNA packaging Genes Cells 2 537–545
F Galibert TM Finan SR, Long et al. (2001) ArticleTitleThe composite genome of the legume symbiont Sinorhizobium meliloti Science 293 668–672 Occurrence Handle1:CAS:528:DC%2BD3MXls1KgtrY%3D Occurrence Handle11474104
H-D Görtz (1996) Symbiosis in ciliates K Hausmann PC Bradbury (Eds) Ciliates: Cells as organisms Gustav Fischer Verlag Stuttgart 441–462
HD Görtz T Brigge (1998) ArticleTitleIntracellular bacteria in protozoa Naturwissenschaften 85 359–368
DC Heruth FR Pond JA Dilts RL Quackenbush (1994) ArticleTitleCharacterization of genetic determinants for R body synthesis and assembly in Caedibacter taeniospiralis 47 and 166 J Bacteriol 176 3559–3567
H Ikeda J Tomizawa (1968) ArticleTitlePhage PI, an extrachromosomal replicator unit Cold Spring Harbor Symp Quant Biol 33 791–798
T Kaneko Y Nakamura S Sato K Minamisawa T Uchiumi S Sasamoto A Watanabe K Idesawa M Iriguchi K Kawashima M Kohara M Matsumoto S Shimpo H Tsuruoka T Wada M Yamada S Tabata (2002) ArticleTitleComplete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110 DNA Res 9 189–197 Occurrence Handle12597275
J Kim J Nietfeldt AK Benson (1999) ArticleTitleOctamer-based genome scanning distinguishes a unique subpopulation of Escherichia coli O157:H7 strains in cattle Proc Natl Acad Sci USA 96 13288–13293
M Kirndorfer A Jager G Klug (1998) ArticleTitleIntegration host factor affects the oxygenregulated expression of photosynthesis genes in Rhodobacter capsulatus Mol Gen Genet 258 297–305
JR Klein GM Dunny (2002) ArticleTitleBacterial group II introns and their association with mobile genetic elements Front Biosci 1 1843–1856
OI Kulaeva EV Koonin JC Wootton AS Levine R Woodgate (1998) ArticleTitleUnusual insertion element polymorphisms in the promoter and terminator regions of the mucAB-like genes of R471a and R446b Mutat Res 397 247–262
J Kusch M Stremmel M Schweikert V Adams HJ Schmidt (2000) ArticleTitleThe toxic symbiont Caedibacter caryophila in the cytoplasm of Paramecium novaurelia Microb Ecol 40 330–335
J Kusch L Czubatinski S Wegmann M Hübner M Alter P Albrecht (2002) ArticleTitleCompetitive advantages of Caedibacter-infected Paramecia Protist 153 47–58
WG Landis (1981) ArticleTitleThe ecology, role of the killer trait, and interactions of five species of the Paramecium aurelia complex inhabiting the littoral zone Can J Zool 59 1734–1743
WG Landis (1987) ArticleTitleFactors determining the frequency of the killer trait within populations of the Paramecium aurelia complex Genetics 115 197–205
AS Lang JT Beatty (2001) ArticleTitleThe gene transfer agent of Rhodobacter capsulatus and “constitutive transduction” in prokaryotes Arch Microbiol 175 241–249
AS Lang TA Taylor JT Beatty (2002) ArticleTitleEvolutionary implications of phylogenetic analyses of the gene transfer agent (GTA) of Rhodobacter capsulatus J Mol Evol 55 534–543
K Lehmann U Schmidt (2003) ArticleTitleGroup II introns: structure and catalytic versatility of large natural ribozymes Grit Rev Biochem Mol Biol 38 249–303
N Linka H Hurka BF Lang G Burger HH Winkler C Stamme C Urbany I Seil J Kusch HE Neuhaus (2003) ArticleTitlePhylogenetic relationships of non-mitochondrial nucleotide transport proteins in bacteria and eukaryotes Gene 306 27–35
AV Lukashin M Borodovski (1998) ArticleTitleGeneMark.hmm: New solutions for gene finding Nucleic Acids Res 26 1107–1115 Occurrence Handle10.1093/nar/26.4.1107 Occurrence Handle1:CAS:528:DyaK1cXhvVWksr4%3D Occurrence Handle9461475
J Maniloff HW Ackermann (1998) ArticleTitleTaxonomy of bacterial viruses: Establishment of tailed virus genera and the order Caudovirales Arch Virol 143 2051–2063
BL Marrs (1974) ArticleTitleGenetic recombination in Rhodopseudomonas capsulata Proc Natl Acad Sci USA 71 971–973
PT Mmolawa CJ Thomas MW Heuzenroeder (2003) ArticleTitleGenomic structure of the Salmonella enterica serovar typhimurium DT64 bacteriophage ST64T: Evidence for modular genetic architecture J Bacteriol 185 3473–3475
M Motallebi-Veshareh DA Rouch CM Thomas (1990) ArticleTitleA family of ATPases involved in active partitioning of diverse bacterial plasmids Mol Microbiol 4 1455–1463
JA Mueller (1963) ArticleTitleSeparation of kappa particles with infective activity from those with killing activity and identification of the infective particles in Paramecium aurelia Exp Cell Res 30 492–508
K Nakayama K Takashima H Ishihara T Shinomiya M Kageyama S Kanaya M Ohnishi T Murata H Mori T Hayashi (2000) ArticleTitleThe R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage Mol Microbiol 38 213–231
K Nelson I Paulsen C Weinel et al. (2002) ArticleTitleComplete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440 Environ Microbiol 4 799–808 Occurrence Handle10.1046/j.1462-2920.2002.00366.x Occurrence Handle1:CAS:528:DC%2BD3sXkvVGrsw%3D%3D Occurrence Handle12534463
F Pond I Gibson J Lalucat RL Quackenbush (1989) ArticleTitleR-Body-producing bacteria Bacteriol Rev 53 25–67
JR Preer SuffixJr LB Preer A Jurand (1974) ArticleTitleKappa and other endobionts in Paramecium aurelia Bacteriol Rev 38 113–163
LB Preer JR Preer SuffixJr (1964) ArticleTitleKilling activity from lysed particles of Paramecium Genet Res 5 230–239
LB Preer A Jurand JR Preer BM Rudman (1972) ArticleTitleThe classes of kappa in Paramecium aurelia J Cell Sci 11 581–600
RL Quackenbush (1977) ArticleTitlePhylogenetic relationships of bacterial endobionts of Paramecium aurelia: Deoxyribonucleotide sequence relationship of 51 kappa and its mutants J Bacteriol 129 895–900
RL Quackenbush (1978) ArticleTitleGenetic relationships among bacterial endobionts of Paramecium aurelia: Polynucleotide sequence relationships among members of Caedibacter J Gen Microbiol 108 181–187
RL Quackenbush (1983) ArticleTitlePlasmids from bacterial endobionts of hump-killer paramecia Plasmid 9 298–306
RL Quackenbush JA Burbach (1983) ArticleTitleCloning and expression of DNA sequences associated with the killer trait of Paramecium tetraurelia stock 47 Proc Natl Acad Sci USA 80 250–254
RL Quackenbush BJ Cox JA Kanabrocki (1986a) Extrachromosomal elements of extrachromosomal elements of paramecia, and their extrachromosomal elements RB Wickner A Hinnebusch AM Lambowitz IC Gunsalus A Hollaender (Eds) Extrachromosomal elements in lower eukaryontes Plenum Press New York 265–278
RL Quackenbush JA Dilts BJ Cox (1986b) ArticleTitleTransposonlike elements in Caedibacter taeniospiralis J Bacteriol 166 349–352
VK Ravin MG Shulga (1970) ArticleTitleEvidence for extrachromosomal location of prophage N15 Virology 40 800–807
TD Read SL Salzberg M Pop M Shumway L Umayam L Jiang E Holtzapple JD Busch KL Smith JM Schupp D Solomon P Keim CM Fraser (2002) ArticleTitleComparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis Science 296 2028–2033
HJ Schmidt H-D Görtz RL Quackenbush (1987) ArticleTitleCaedibacter caryophilus sp. nov., a killer symbiont inhabiting the macronucleus of Paramecium caudatum Int J Syst Bacteriol 37 459–462
HJ Schmidt H-D Görtz FR Pond RL Quackenbush (1987b) ArticleTitleCharacterization of Caedibacter endonucleosymbionts from the macronucleus of Paramecium caudatum and the identification of a mutant with blocked R body synthesis Exp Cell Res 174 49–57
K Shiraishi K Hanada Y Iwakura H Ikeda (2002) ArticleTitleRoles of RecJ, RecO, and RecR in RecET-mediated illegitimate recombination in Escherichia coli J Bacteriol 184 4715–4721s
E Strauch H Kaspar C Schaudinn P Dersch K Madela C Gewinner S Hertwig J Wecke B Appel (2001) ArticleTitleCharacterization of enterocoliticin, a phage tail-like bacteriocin, and its effect on pathogenic Yersinia enterocolitica strains Appl Environ Microbiol 67 5634–5642
C Vander Byl AM Kropinski (2000) ArticleTitleSequence of the genome of Salmonella bacteriophage P22 J Bacteriol 182 6472–6481
DH Walker SuffixJr JT Walker (1975) ArticleTitleGenetic studies of coliphage P1. I. Mapping by use of prophage deletions J Virol 16 525–534
HH Winkler HE Neuhaus (1999) ArticleTitleNon-mitochondrial ATP transport TIBS 24 64–68
HC Yen NT Hu BL Marrs (1979) ArticleTitleCharacterization of the gene transfer agent made by an overproducer mutant of Rhodopseudomonas capsulata J Mol Biol 131 157–168
K Yokoyama K Makino Y Kubota M Watanabe S Kimura CH Yutsudo K Kurokawa K Ishii M Hattori H Abe T Iida K Yamamoto T Hayashi T Yasunaga T Honda C Sasakawa H Shinagawa (2000) ArticleTitleComplete nucleotide sequence of the prophage VT1-Sakai carrying the Shiga toxin 1 genes of the enterohemorrhagic Escherichia coli O157:H7 strain derived from the Sakai outbreak Gene 258 127–139
Zhang Y, Muyrers JPP, Rientjes J, Stewart AF (2003) Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol Biol 4: art.no. 1
L Zheng R Halberg S Roels H Ichikawa L Kroos R Losick (1992) ArticleTitleSporulation regulatory protein GerE from Bacillus subtilis binds to and can activate or repress transcription from promoters for mother-cell-specific genes J Mol Biol 226 1037–1050
T Zhou S Radaev BP Rosen DL Gatti (2000) ArticleTitleStructure of the ArsA ATPase: the catalytic subunit of a heavy metal resistance pump EMBO J 19 4838–45
Acknowledgments
We thank S. Fokin and L. Preer for the supply of Paramecium cultures. We are grateful for helpful comments by an anonymous reviewer. This research was funded in part by a grant from the Deutsche Forschungsgemeinschaft to J.K. (DFG KU 1237).
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Debashish Bhattacharya
Rights and permissions
About this article
Cite this article
Jeblick, J., Kusch, J. Sequence, Transcription Activity, and Evolutionary Origin of the R-BodyCoding Plasmid pKAP298 from the Intracellular Parasitic BacteriumCaedibacter taeniospiralis. J Mol Evol 60, 164–173 (2005). https://doi.org/10.1007/s00239-004-0002-2
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0002-2