
Abstract
The 1.709 or satellite IV repeated DNA family originally isolated from the domestic cow was analyzed using Southern blotting, pulsed field gel electrophoresis, fluorescence in situ hybridization, and DNA sequencing in species belonging to the genera Bos, Bison, Bubalus, Syncerus, Boselaphus, and Tragelaphus. Hybridization indicates that the family has been amplified in Bos, Bison, Bubalus, and Syncerus but not in Boselaphus or Tragelaphus. Pericentromeric, higher-order repeat substructure exists in all species, with multimeric arrays ranging in size from 10 to 1500 kb. Sequence analysis of a 492-bp PCR product revealed comparable levels (0.2–4.5%) of intra- and interspecific divergence when species of Bos and Bison were compared, supporting the idea that species of these two genera should be recognized under the genus Bos. Alternatively, all Syncerus sequences cluster as a monophyletic group on an evolutionary tree and differ from those of Bos/Bison by about 13%. Comparing these findings with the fossil record indicates that concerted evolution has occurred since Bos/Bison and Syncerus last shared a common ancestor (5.0 MYA) but before the radiation of the genus Bos (2.5 MYA): GenBank accession numbers AY517856-AY517904.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The comparative study of satellite DNA families among closely related species remains a fruitful enterprise since tremendous variation exists regarding the extent of sequence conservation (Mravinac et al. 2002; Nijman and Lenstra 2001), the degree of concerted evolution (Elder and Turner 1994; Modi 1993b), chromosomal distribution (Li et al. 2000; Modi 1993a; Modi et al. 1996), and higher-order repeat (HOR) structure (Warburton et al. 1996). One taxonomic group of mammals extremely well suited for the evolutionary analyses of satellite DNAs is the artiodactyls. This collection of over 250 species of even-toed ungulates occurs throughout the world, contains a large number of structurally complex satellite families (Li et al. 2000; Pech et al. 1979; Poschl and Streeck 1980; Skowronski et al. 1984), exhibits substantial interspecific chromosomal variation (Gallagher and Womack 1992), and presents a number of interesting molecular evolutionary questions (Robinson et al. 2000).
The 1.709 or satellite IV family isolated from the domestic cow (Skowronski et al. 1984) is found only among closely related species in the tribe Bovini, including cattle, bison, and buffaloes (Jobse et al. 1995; Modi et al. 1996; Nijman and Lenstra 2001). It was further shown that related species revealed conserved hybridization (Jobse et al. 1995; Modi et al. 1996; Nijman and Lenstra 2001), that limited interspecific sequence variation exists (Nijman and Lenstra 2001), and that the repeat has a pericentromeric chromosomal distribution (Modi et al. 1996). The present study extends these earlier observations in two major respects. First, the nucleotide sequence of a 492-bp region was determined and phylogenetically analyzed for 49 recombinant clones from eight species or subspecies. Further, pulsed field gel electrophoresis (PFGE) was carried out over two genomic size ranges. These data reveal the pattern of concerted evolution both within and between species and indicate that homogenized arrays of HORs characterize the organization of this centromeric satellite family.
Materials and Methods
Sample Material
Fibroblast cell lines were available from 11 taxa of artiodactyls: tribe Bovini—Bos taurus (domestic or taurus cow), Bos indicus (zebu or indicus cow), Bos gaurus (gaur; two individuals), Bos javanicus (banteng), Bison bison (American bison), Bison bonasus (wisent), Bubalus bubalis (Asian river buffalo), Syncerus caffer caffer (African cape buffalo), and Syncerus caffer nanus (African Congo buffalo); tribe Boselaphini—Boselaphus tragelaphus (nilgai); and tribe Tragelaphini—Tragelaphus imberbis (lesser kudu). See Gallagher and Womack (1992) and Modi et al. (1996) for information on sample origin.
PCR, Cloning, and Sequencing
Repetitive DNA was sequenced from 8 of the 11 taxa listed above, all except Asian river buffalo, nilgai, and lesser kudu. Sequencing templates were obtained by cloning PCR products. PCR was carried out in a 20-µl volume containing 25 ng of DNA, 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, a 200 µM concentration of each dNTP, 1 pmol of each primer (forward, TTTACCTT AGAACAAACCGAGGCAC; and reverse, TACGGAAAGGAA AGATGACCTGACC), and 0.001 units of Taq Gold (Perkin Elmer) polymerase. This primer pair amplifies a 536-bp product from nucleotide positions 33 to 569 (Modi et al. 1993) in the reference sequence GenBank X00979. This region of the cow repeat was chosen since it lacks short, repetitive sequences and was expected to be informative in other species. It should be noted that primer selection may impose a bias, since only sequences homologous to the primer sites will be amplified. Thermal cycling included an initial 94°C denaturation step for 10 min, followed by 35 cycles of 94°C denaturation for 30 s, 58°C annealing for 1 min, and a 72°C extension step for 1 min.
PCR products were cloned into alkaline phosphatase-treated Bluescript KS+ vector (Stratagene). XL-I Blue bacteria (Stratagene) were transformed, and plasmid DNA was purified using Wizard mini-prep DNA purification kits (Promega). Inserts were sequenced in both directions using T3/T7 primers and dideoxy chain termination kits, and reactions run on a 373A DNA sequencing system (ABI). Sequences were aligned and intersequence divergence was calculated using the program MEGA2 (Kumar et al. 2001). Phylogenetic trees were constructed using PAUP4.0b10 (Swofford 2001).
Filter Hybridization
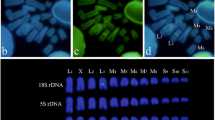
For Southern blotting, 2 µg of genomic DNA was digested with HincII, transferred to a nylon membrane, and hybridized with a 32P α-dCTP-labeled 536-bp PCR product from the domestic cow. Posthybridization washes were in 0.1× SSC, 0.5% SDS at 50°C for 30 min (additional details given by Modi et al. [1993]). PFGE was carried out on genomic DNA in agarose plugs derived from imbedded fibroblast cells. The rare cutting ApaI, ClaI, MluI, SalI, and XhoI restriction enzymes were used. Digested DNAs were subjected to electrophoresis under two gel conditions, permitting resolution in two size ranges (Fig. 1B–D). The hybridization probe and conditions were the same as those used in Southern blotting (PFGE details given by Ivanov and Modi [1996]).

Filter hybridization of bovid DNAs after probing with a 32P α-dCTP-labeled 536-bp PCR product from the taurus cow 1.709 repeat. A Southern blot following HincII digestion. The 3.8-kb monomer (1°) is indicated in the taurus and indicus cattle lanes. B–D Pulsed field gel electrophoresis following digestion with a series of rare-cutting restriction enzymes. Arrows highlight discrete restriction fragments, representing higher-order repeats.
Fluorescence In Situ Hybridization (FISH)
Metaphase chromosomes were harvested from primary fibroblast cell lines from four species: African cape buffalo, Asian river buffalo, American bison, and nilgai. FISH was performed using a biotin-11-dUTP-labeled 536-bp PCR product from the domestic cow as hybridization probe. Posthybridization washes were in 50% formamide in 2× SSC at 39°C for 15 min. Details of the cell culture and FISH procedures have been reported (Gallagher and Womack 1992; Modi et al. 1996).
Results
Southern Blotting
HincII-digested genomic DNA from 10 taxa is shown in Fig. 1A. Hybridization was absent to nilgai and lesser kudu. The remaining eight taxa revealed three different restriction patterns. Taurus and indicus cattle share a common pattern, with a full-length 3.8-kb monomer and another fragment of about 1.8 kb. Gaur, banteng, bison, and wisent share a second pattern where they also have the 1.8-kb band in addition to a fragment of about 500 bp. Further, the bison has a band of about 2.5 kb in size. The Congo and Cape buffaloes present the third pattern, with a doublet just under 6 kb and another restriction fragment about 2.5 kb in size.
Pulsed Field Gel Electrophoresis
DNA from all species contained smears in addition to discrete bands ranging from less than 10 to over 1500 kb (Fig. 1B–D). The large smears suggest that much of the satellite is contained in arrays with restriction sites spaced at irregular intervals, while the discrete bands represent HORs where restriction sites occur at regular intervals. In many cases multiple bands are observed in a single lane (i.e., SalI digest of gaur; Fig. 1D). Interspecific differences in restriction patterns were noted. Cattle, gaur, banteng, bison, and wisent (belonging to the genus Bos or Bison) revealed patterns generally similar to each other and different from those of Congo (Syncerus) and river (Bubalus) buffaloes. The primary difference was that Congo and river buffalo DNA was digested to a greater extent with certain enzymes. For example ClaI, MluI, and SalI digested buffalo DNAs below 500 kb in size, while much of the DNA from the species of Bos and Bison remained higher in molecular weight (Fig. 1B and D). Also, interspecific differences in the sizes of discrete bands were noted for many enzymes (Fig. 1B–D). This is clearly seen for the SalI and XhoI digests in the bison, wisent, and Congo buffalo (Fig. 1B).
Fluorescence In Situ Hybridization
Metaphase chromosomes from four species are presented in Fig. 2. Hybridization patterns in the African cape buffalo, Asian river buffalo, and American bison were very similar, with light to moderate signal observed at the centromeres of 50–75% of the chromosomes in each species (Fig. 2A–C). Hybridization was absent to chromosomes of the nilgai (Fig. 2D), consistent with results of the Southern blotting.

Fluorescence in situ hybridization of metaphase chromosomes following probing with biotin-11-dUTP-labeled 536-bp PCR product from the taurus cow 1.709 repeat. A African cape buffalo (Syncerus caffer caffer), 2n = 52, four pairs of biarmed autosomes. B Asian river buffalo (Bubalus bubalis), 2n = 50, five pairs of biarmed autosomes (chromosomes from two adjacent cells are present). C American bison (Bison bison), 2n = 60, no biarmed autosomes. D Nilgai (Boselaphus tragocamelus), 2n = 46, six pairs of biarmed autosomes.
Sequence Analyses
Forty-nine clones from nine individuals were sequenced (Supplement 1). One hundred thirty-one of the 492 sites were polymorphic, 89 of which were phylogenetically informative. These results considerably extend those of Nijman and Lenstra (2001), who sequenced PCR products of nearly the same region. In their seven sequences, they found 22 of 461 sites to be polymorphic, 19 of which were variable here. The greater variability found here is presumably due to the much larger number of sequences examined. Pairwise comparisons of all 49 sequences enabled the calculation of intraspecific and interspecific nucleotide divergence. Within-species levels of divergence ranged from 0.2 to 4.7% (Supplement 2). Indicus cow and American bison had slightly lower levels than did the other taxa. Interspecific levels of divergence among the six species of Bos and Bison are similar to intraspecific levels among these same species (0.2–4.5%), while the sequences of Bos and Bison are 10–15% divergent from those of Syncerus.
Phylogenetic trees constructed using parsimony or neighbor joining (Fig. 3) produced similar topologies. Sequences from the African cape and Congo buffaloes clustered together in a monophyletic group with 99% bootstrap support, distinct from those of Bos and Bison. Furthermore, sequences from the latter two genera group together irrespective of their species of origin.

Neighbor-joining tree of 49 sequences, using the Kimura two-parameter distance measure, constructed by PAUP. Bootstrap support for the branch leading to the Congo and cape buffaloes is 99%,while none of the branches separating the major clades of Bos and Bison had more than 60% support.
Discussion
Genomic Organization of Higher-Order Repeats
Southern blotting revealed only two or three distinct bands in every species, suggesting relative homogeneity of the individual monomers within a genome with respect to the distribution of HincII sites. These results are similar to those reported for several of these species using different enzymes (Jobse et al. 1995; Modi et al. 1993, 1996). Further, species of Bos (gaur, banteng) and Bison (bison, wisent) have similar restriction patterns, suggesting that the two genera should be synonymized (see systematic discussion below). The structural organization of this repetitive family has been determined only from the domestic cow, where a 3.8-kb monomer was defined and found to be mosaic in structure, containing sequence unique to the satellite IV family in addition to segments of two bovine interspersed repeats (Skowronski et al. 1984). Since the hybridization probe used here contained unique sequence from the 5′ end of the cow monomer, our discussion of the material present in other species is limited to this region. The exact sizes of the monomers, and whether or not they are mosaic in structure in other species, remains to be determined.
The PFGE revealed HORs of various sizes (10–1500 kb) within and between species. These HORs are restriction fragments containing tandemly arrayed monomers, presumably with varying levels of sequence divergence between monomers. These multimeric arrays are delimited by the presence of a rare-cutting restriction site. Since multiple bands were observed in a single lane of a gel, HORs of different sizes exist within a genome. As this satellite family is dispersed to multiple chromosomes in these species, it is possible that different HORs are localized on different chromosomes as shown for human α-satellite (Willard 1985).
Sequence Homogenization and Interspecific Systematics
Sequence analyses revealed extensive similarity between species in the genera Bos and Bison. In fact, intraspecific divergence (0.2 to 4.5%) was equal to that distinguishing species. This may be contrasted with the sequence differentiation between Bos/Bison and Syncerus (10 to 15%). Previous systematic analyses recognized Syncerus as an outgroup to Bos and Bison, and species in the latter two genera are so closely related that no consensus opinion of their relationships exists (Groves 1981; Miyamoto et al. 1989; Modi et al. 1996; Wall et al. 1992), such that Groves (1981) recognized all members under the genus Bos. Paleontological dating contends that Syncerus and Bos (including Bison) shared a common ancestor about 5.0 MYA, and recent species of Bos 2.5–1.0 MYA (Groves 1981). Evaluating the present sequence data in the context of the fossil record enables us to geologically date the timing and extent of molecular homogenization (concerted evolution) that has occurred. Since the six species of Bos/Bison exhibit levels of divergence within species that overlap those between species, it is evident that genome-wide homogenization has not occurred within any of these species since they shared a common ancestor. However, as the sequences from Syncerus cluster as a monophyletic group distinct from those of Bos/Bison, homogenization has occurred in the Syncerus and/or Bos/Bison lineages since they last shared a common ancestor (between 5 and 2.5 MYA). To emphasize that general patterns regarding the timing and extent of concerted evolution do not exist when considering different taxonomic groups, the present findings may be contrasted with two other reports. Among tenebrionid beetles that diverged 60–50 MYA, members of the PRAT satellite DNA family from different species are indistinguishable in nucleotide sequence (Mravinac et al. 2002). On the other hand, concerted evolution has taken place at the level of the population in pupfish satellite DNA, where homogenized arrays differed little within populations but markedly between populations (Elder and Turner 1994).
References
Jr JF Elder BJ Turner (1994) ArticleTitleConcerted evolution at the population level: Pupfish HindIII satellite DNA sequences. Proc Natl Acad Sci USA 91 994–998 Occurrence Handle1:CAS:528:DyaK2cXht1Kksbo%3D Occurrence Handle8302879
Jr DS Gallagher JE Womack (1992) ArticleTitleChromosome conservation in the Bovidae. J Hered 83 287–298 Occurrence Handle1401875
CP Groves (1981) ArticleTitleSystematic relationships in the Bovini (Artiodactyla, Bovidae). Z Zool Syst Evolut-forsch 19 264–278
SV Ivanov WS Modi (1996) ArticleTitleMolecular characterization of the complex sex-chromosome heterochromatin in the rodent Microtus chrotorrhinus. Cytogenet Cell Genet 75 49–56 Occurrence Handle1:CAS:528:DyaK2sXmtVGgtA%3D%3D Occurrence Handle8995489
C Jobse JB Buntjer N Haagsma HJ Breukelman JJ Beintema JA Lenstra (1995) ArticleTitleEvolution and recombination of bovine DNA repeats. J Mol Evol 41 277–283 Occurrence Handle1:CAS:528:DyaK2MXnslWntrY%3D Occurrence Handle7563113
S Kumar K Tamura IB Jakobsen M Nei (2001) ArticleTitleMEGA2: Molecular evolutionary genetics analysis software. Bioinformatics 17 1244–1245 Occurrence Handle1:CAS:528:DC%2BD38XmtVCktQ%3D%3D Occurrence Handle11751241
YC Li C Lee TH Hseu SY Li CC Lin TH Hsu (2000) ArticleTitleDirect visualization of the genome distribution and organization of two cervid centromeric satellite DNA families. Cytogenet Cell Genet 89 192–198 Occurrence Handle1:CAS:528:DC%2BD3cXmsF2msLw%3D Occurrence Handle10965121
MM Miyamoto SM Tanhauser PJ Laipis (1989) ArticleTitleSystematic relationships in the artiodactyl tribe Bovini (family Bovidae), as determined from mitochondrial DNA sequences. Syst Zool 38 342–349
WS Modi (1993a) ArticleTitleComparative analyses of heterochromatin in Microtus: Sequence heterogeneity and localized expansion and contraction of satellite DNA arrays. Cytogenet Cell Genet 62 142–148 Occurrence Handle1:CAS:528:DyaK3sXkvFajurY%3D
WS Modi (1993b) ArticleTitleHeterogeneity in the concerted evolution process of a tandem satellite array in meadow mice (Microtus). J Mol Evol 37 48–56 Occurrence Handle1:CAS:528:DyaK3sXkvFaju7c%3D
WS Modi DS Gallagher JE Womack (1996) ArticleTitleMolecular organization and chromosomal localization of six highly repeated DNA families in the bovine genome. Anim Biotech 4 143–161
WS Modi DS Gallagher JE Womack (1993) ArticleTitleEvolutionary histories of highly repeated DNA families among the Artiodactyla (Mammalia). J Mol Evol 42 337–349
B Mravinac M Plohl N Mestrovic D Ugarkovic (2002) ArticleTitleSequence of PRAT satellite DNA “frozen” in some coleopteran species. J Mol Evol 54 774–782 Occurrence Handle10.1007/s0023901-0079-9 Occurrence Handle1:CAS:528:DC%2BD38XkvFOmtL4%3D Occurrence Handle12029359
IJ Nijman JA Lenstra (2001) ArticleTitleMutation and recombination in cattle satellite DNA: A feedback model for the evolution of satellite DNA repeats. J Mol Evol 52 361–371 Occurrence Handle1:CAS:528:DC%2BD3MXivVSisLk%3D Occurrence Handle11343132
M Pech RE Streeck HG Zachau (1979) ArticleTitlePatchwork structure of a bovine satellite DNA. Cell 18 883–893 Occurrence Handle1:CAS:528:DyaL3cXjs1ajtw%3D%3D Occurrence Handle519761
E Poschl RE Streeck (1980) ArticleTitlePrototype sequence of bovine 1.720 satellite DNA. J Mol Biol 143 147–153 Occurrence Handle1:STN:280:Bi6D2s3jtFU%3D Occurrence Handle7441759
TJ Robinson O Wittekindt JJ Pasantes WS Modi W Schempp DJ Morris-Rosendahl (2002) ArticleTitleStable methylation patterns in interspecific antelope hybrids and the characterization and localization of a satellite fraction in the Alcelaphini and Hippotragini. Chromosome Res 8 635–643 Occurrence Handle10.1023/A:1009294226213
J Skowronski A Plucienniczak A Bednarek J Jaworski (1984) ArticleTitleBovine 1.709 satellite. Recombination hotspots and dispersed repeated sequences. J Mol Biol 177 399–416 Occurrence Handle1:CAS:528:DyaL2MXhvVKmsQ%3D%3D Occurrence Handle6088777
D Swofford (2001) PAUP*, phylogenetic analysis using parsimony. Sinauer Associates Sunderland, MA
AD Wall SK Davis BM Read (1992) ArticleTitlePhylogenetic relationships in subfamily bovinae (Mammalia–Artiodactyla) based upon ribosomal DNA. J Mammal 73 262–275
PE Warburton T Haaf J Gosden D Lawson HF Willard (1996) ArticleTitleCharacterization of a chromosome-specific chimpanzee alpha satellite subset: Evolutionary relationship to subsets on human chromosomes. Genomics 33 220–228 Occurrence Handle10.1006/geno.1996.0187 Occurrence Handle1:CAS:528:DyaK28Xis1ensb8%3D Occurrence Handle8660971
HF Willard (1985) ArticleTitleChromosome-specific organization of human alpha satellite DNA. Am J Hum Genet 37 524–532 Occurrence Handle1:CAS:528:DyaL2MXks1Gqurk%3D Occurrence Handle2988334
Acknowledgements
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This work was funded in part by NCI, NIH Contract No. NO1-CO-124000. Supplementary material is available.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Modi, W.S., Ivanov, S. & Gallagher, D.S. Concerted Evolution and Higher-Order Repeat Structure of the 1.709 (Satellite IV) Family in Bovids . J Mol Evol 58, 460–465 (2004). https://doi.org/10.1007/s00239-003-2567-6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2567-6