
Abstract
Molecular investigations of deep-level relationships within and among the animal phyla have been hampered by a lack of slowly evolving genes that are amenable to study by molecular systematists. To provide new data for use in deep-level metazoan phylogenetic studies, primers were developed to amplify a 1.3-kb region of the α subunit of the nuclear-encoded sodium–potassium ATPase gene from 31 bilaterians representing several phyla. Maximum parsimony, maximum likelihood, and Bayesian analyses of these sequences (combined with ATPase sequences for 23 taxa downloaded from GenBank) yield congruent trees that corroborate recent findings based on analyses of other data sets (e.g., the 18S ribosomal RNA gene). The ATPase-based trees support monophyly for several clades (including Lophotrochozoa, a form of Ecdysozoa, Vertebrata, Mollusca, Bivalvia, Gastropoda, Arachnida, Hexapoda, Coleoptera, and Diptera) but do not support monophyly for Deuterostomia, Arthropoda, or Nemertea. Parametric bootstrapping tests reject monophyly for Arthropoda and Nemertea but are unable to reject deuterostome monophyly. Overall, the sodium–potassium ATPase α-subunit gene appears to be useful for deep-level studies of metazoan phylogeny.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent investigations of the relationships within and among the metazoan (multicellular animal) phyla have employed morphological data (e.g., Eernisse et al. 1992; Nielsen et al. 1996), molecular data (e.g., Eeckhaut et al. 2000; Field et al. 1988; Giribet and Ribera 1998; Ruiz-Trillo et al. 1999), and combined morphological and molecular data sets (Giribet et al, 2000; Giribet et al. 2001; Zrzavy et al. 1998). Most molecular investigations of metazoan phylogeny to date have utilized the nuclear small-subunit (18S) ribosomal RNA gene. Analyses of 18S data have provided a number of important insights into metazoan relationships at several levels, including evidence supporting a clade of molting animals (e.g., nematodes and arthropods) dubbed Ecdysozoa (Aguinaldo et al. 1997). However, some aspects of metazoan phylogeny remain unclear, most notably relationships within Lophotrochozoa, a clade that includes Annelida, Mollusca, and several other phyla (Halanych 1998; Halanych et al. 1995).
To address these remaining ambiguities, some researchers have studied large multilocus data sets from representatives of a few taxa (those for which complete genomic sequences or large EST databases are available) (e.g., Blair et al. 2002; Mushegian et al. 1998; Wang et al. 1999), while others have attempted to gather sequences for one or a few genes from representatives of many taxa (e.g., Giribet et al. 2001). Both approaches have merit, but at this time large sequence databases exist for only a few metazoans, and most molecular systematists simply do not have the resources to generate complete genomic sequences (or EST profiles) for representatives of several metazoan taxa. Therefore, PCR-based studies of relatively small numbers of genes will continue to be the predominant approach employed by most molecular systematists, because it allows several taxa of interest to be studied in a cost-effective manner.
Other approaches to investigating phylogeny beyond comparisons of nucleotide substitutions within one or more genes are also being explored. Rare genomic changes (RGCs) such as intron indels, SINEs and LINEs, mitochondrial and nuclear genetic code variants, and gene duplications may also play an important role in resolving phylogenetic problems at various levels, including relationships among and within the animal phyla (Rokas and Holland 2000). Thus far, three types of RGC have been used to investigate aspects of metazoan phylogeny: mtDNA code variants (Castresana et al. 1998), signature Hox gene motifs (de Rosa et al. 1999), and mtDNA gene order (Boore 1999). Of these, mtDNA gene order has received the most attention.
Although studies of mitochondrial gene order and complete mitochondrial genome sequences seem to corroborate findings based on analyses of 18S data alone (Boore and Brown 2000; Boore et al. 1998; von Nickisch-Rosenegk et al. 2001), this line of research seems to have produced few novel insights to date (but see Boore and Staton 2002). This is due in part to the difficulty and expense of sequencing complete mitochondrial genomes. Mitochondrial gene order rearrangements initially seemed to be an ideal source of information for studying ancient divergences, but the presumed rarity of mitochondrial gene rearrangement events has been called into question recently (Dowtin and Austin 1999; Flook et al. 1995; Hickerson and Cunningham 2000; Le et al. 2000; Mindell et al. 1998; Rawlings et al. 2001; Shao et al. 2001a,b), and the development of appropriate methods for analyzing gene order data has been difficult (Blanchette et al. 1999). Complete mitochondrial sequence data seem promising, but even 14+ kb of mitochondrial protein-coding sequence is insufficient for resolving relationships among basal vertebrates (Cao et al. 1998), suggesting that the ability of these data to resolve interphylum relationships also may be limited. These observations suggest that mitochondrial data alone may be insufficient for the resolution of persistent problems in deep-level animal phylogeny—additional data from the nuclear genome, in the form of either sequences of additional nuclear genes from representatives of several phyla or new RGCs (or both) will be necessary.
A few studies have focused on nuclear genes other than 18S, most notably the large-subunit (28S) ribosomal rRNA gene (Christen et al. 1991; Mallatt and Winchell 2002) and the gene that encodes elongation factor 1-α (EF-1α) (Kobayashi et al. 1996; Kojima 1998; Kojima et al. 1993; McHugh 1997; Regier and Shultz 1997, 1998). Unfortunately, the value of the EF-lα gene for elucidating relationships among phyla may be somewhat limited (Littlewood et al. 2001), although it has recovered some interesting patterns, including paraphyly of Annelida with respect to Echiura and Pogonophora (McHugh 1997) and paraphyly of Crustacea with respect to Hexapoda (Regier and Shultz 1997). Comparative studies of a few other nuclear genes have also been published, including studies of aldolase and triose phosphate isomerase (Nikoh et al. 1997), intermediate filament proteins (Erber et al. 1998), lamins (Erber et al. 1999), heat shock proteins (Borchiellini et al. 1998), protein kinases (Kruse et al. 1998), elongation factor-2 (Regier and Shultz 2001), and myosin heavy chain type II (Ruiz-Trillo et al. 2002). Thus far, however, these data sets have been very limited in taxonomic scope, likely reflecting the difficulty of designing “universal” PCR primers that can amplify a particular gene region from representatives of most (or all) metazoan phyla.
Studies of metazoan relationships have clearly been hampered by a relative lack of slowly evolving genes that can be studied using standard laboratory techniques available to most molecular systematists (e.g., PCR). Unfortunately, little exploratory work has been done to develop additional nuclear protein-coding gene markers for studies of deep intraphylum and interphylum relationships within Metazoa. Friedlander et al. (1992) described 14 promising candidate genes for phylogenetic research within Metazoa and listed EF-1α, elongation factor-2, Na+/K+ ATPase, enolase, and glucose-6-phosphate dehydrogenase as genes that may be suitable for use in deep-level phylogenetic studies of metazoans. In an effort to develop new nuclear-protein coding markers to address remaining questions in inter- and intraphylum metazoan relationships, we developed three pairs of oligonucleotide primers for amplification of a 1.3-kb region of coding sequence from the gene encoding the α subunit of Na+/K+ ATPase (the “sodium pump” enzyme). We used these primers to amplify this gene region from mRNA extracts from representatives of a diverse group of thirty-one bilaterian (bilaterally symmetrical) metazoans. These sequences were used to reconstruct relationships among and within several important metazoan taxa and to evaluate the potential value of this gene region for future studies of metazoan phylogeny.
Materials and Methods
Primer Design
An alignment of several metazoan ATPase α-subunit amino acid sequences downloaded from GenBank was visually scanned for short conserved motifs of seven or more amino acids that could serve as PCR primer sites. Oligonucleotides matching five conserved regions were synthesized for use in RT-PCR and PCR (Table 1). The primers have several degenerate sites and are thus unsuitable for use in cycle sequencing reactions. To allow PCR fragments to be directly sequenced, all primers were synthesized with M13 linker sequences attached to their 5′ ends, following Cho et al. (1995) and Regier and Schulz (1997). The 5′-most and 3′-most primers (fATPa and rATPc) were also synthesized without M13 linkers; this primer pair was used in initial RT-PCR amplifications.
RT-PCR and Nested PCR Amplification
Total RNA was extracted from 50–100 mg of tissue using TRI reagent (Molecular Research Center) following the manufacturer’s protocols. The taxa sampled, along with collection locality and GenBank accession numbers, are listed in Table 2. RT-PCR amplification was performed using Ready-To-Go RT-PCR Beads (Amersham Pharmacia Biotech) and the following cycling parameters: 42°C for 20 min (1 cycle), 95°C for 5 min (1 cycle), 95°C for 30 s—38°C for 30 s—72°C for 1 min (32 cycles), 72°C for 7 min (1 cycle). RT-PCR reagent concentrations followed the manufacturer’s suggestions, and a poly (T) primer was used to enhance mRNA amplification. To produce smaller overlapping fragments suitable for direct sequencing, nested PCR reamplifications of the RT-PCR product were performed using the primer pairs fATPa–rATPa, fATPb–rATPb, and fATPc–rATPc (Table 1). Reamplifications were performed under a variety of cycling parameters and reagent concentrations. A regime consisting of 94°C for 90 s (1 cycle), 94°C for 30 s—50°C for 30 s—72°C for 1 min (10 cycles), 94°C for 30 s—44°C for 30 s—72°C for 1 min (25 cycles), followed by a single terminal extension step (72°C for 7 min) on a Perkin–Elmer Gene Amp 9700 was usually satisfactory. Each PCR product was gel-purified using Centri-Sep spin columns (Princeton Separations) and the purified fragment was directly sequenced in both directions using dye terminator chemistry (BigDye Terminators; Applied Biosystems) with M13 sequencing primers. Sequencing reactions were run out on an ABI 373XL automated sequencer. The resulting sequences were assembled and edited in Sequencher (GeneCodes). Edited sequences were aligned using ClustalX (Thompson et al. 1997) with modification by eye in Se-Al (Rambaut 1995) and MacClade 4.03 (Maddison and Maddison 2001).
MP, ME, ML, and Bayesian Analyses
The nucleotide data alignment was analyzed under maximum parsimony (MP; equal weights and implicit weights), minimum evolution (ME; LogDet + invariant distances, using an ML estimate of the proportion of invariant sites), and maximum likelihood (ML) criteria in PAUP* 4.0 (Swofford 2002) and using a Bayesian inference approach in MrBayes 2.0 (Huelsenbeck and Ronquist 2001). Several ATPase α-subunit coding sequences were included with the taxa sampled in this study to increase the diversity of taxa represented (Table 2). Two taxon sets were analyzed—a large taxon set including 24 vertebrate ATPase α-subunit sequences (representing several isoforms) and a smaller taxon set including only 13 vertebrate isoform 1 sequences, each with all available nonvertebrate metazoan ATPase sequences. Two regions of the data set (corresponding to amino acid positions 423–429 and 509–519 in the Drosophila melanogaster sequence) were difficult to align with confidence; these regions were excluded from all analyses. Third codon position sites were also excluded from all nucleotide analyses due to lack of base frequency homogeneity across all sequences (see below). The nucleotide data were also translated to produce an amino acid data matrix, which was analyzed using MP and ML. All MP and ME heuristic searches were performed using 200 random addition sequence replicates with tree bisection–reconnection (TBR) branch-swapping, holding 10 trees at each addition step.
The MP trees were used to evaluate nucleotide substitution models for ML analysis. Likelihood scores were calculated for one MP tree for each taxon set under 56 substitution models, and ModelTest 3.0 (Posada and Crandall 1998) was used to select an appropriate model via a series of hierarchical likelihood ratio tests. The general time-reversible (GTR) DNA substitution model with among-site rate heterogeneity modeled using a mixed-model γ + invariant sites distribution (Γ + I) was chosen by ModelTest as the most adequate model for both taxon sets. Investigation of the likelihood score output revealed that a GTR + Γ + I submodel not evaluated by ModelTest was not significantly worse than a full GTR + Γ + I model (likelihood-ratio test; p = 0.414427 for the small taxon set, p = 0.223130 for the large taxon set). This GTR submodel employed four (rather than six) relative rate parameters: one for A–C transversions and A–G transitions, one for A–T and C–G transversions, one for C–T transitions, and one for G–T transversions (the PAUP* option used was “RCLASS = (a a b b c d)”). To reduce computational time and sampling error, this simpler model was used for all ML searches. To further reduce analysis time, a “successive approximations” approach (e.g., Anderson 2000; Frati et al. 1997) was used. All substitution model parameters were estimated from the data on five starting trees—two MP trees (if more than two MP trees were produced, two very different MP trees were chosen to maximize topological diversity in the starting tree pool), a successive weighting parsimony tree, an implicit weights parsimony tree (k = 2) and a LogDet + I tree. In cases where only one MP tree was found in initial searches, two random trees were also used as starting points for ML searches. Each of these trees (with model parameter estimates fixed) was then used as a starting tree for a round of nearest-neighbor interchange (NNI) branch swapping under ML, followed by reestimation of model parameters and a subsequent round of TBR branch swapping. For each taxon set, all starting points converged on the same ML topology; this tree was accepted as the ML tree. The WAGf+Γ+I substitution model (Whelan and Goldman 2001) was selected as the best-fitting model for the amino acid data matrix following a series of likelihood-ratio tests. TREE-PUZZLE (Schmidt et al. 2002; Strimmer and von Haeseler 1996) was used to perform a maximum likelihood quartet puzzling analysis of the amino acid matrix under this model, with amino acid frequencies, the Γ shape parameter (α), and the proportion of invariant sites (I) estimated from the data.
Nodal support was estimated using equal-weights parsimony bootstrap analysis (100 pseudoreplicates, each consisting of a heuristic search using 100 random addition sequence replicates), maximum likelihood bootstrap analysis using the GTR+Γ+I submodel described above (100 pseudoreplicates generated with SEQBOOT in PHYLIP and analyzed using a successive approximations approach in PAUP*) (Felsenstein 1985, 1995), Bayesian analysis (1 million generations, with the first 200,000 generations discarded as burn-in) (Huelsenbeck and Ronquist 2001), and quartet puzzling (for the amino acid data). The ML submodel used in PAUP* for analysis of the nucleotide data is not available in MrBayes; a standard GTR+Γ+I model was used instead.
Hypothesis Testing
A parametric bootstrapping approach (Hillis et al. 1996; Huelsenbeck et al. 1996) was used to test the level of support in the small taxon set for monophyly of three higher taxa—Deuterostomia, Arthropoda, and Nemertea. Model trees for each hypothesis (deuterostome monophyly, arthropod monophyly, and nemertean monophyly) were constructed by performing ML analyses in PAUP* using the GTR+Γ+I submodel described above and topological constraints to enforce monophyly for each group of interest. One hundred data sets were simulated on each model tree with Seq-Gen 1.25 (Rambaut 1995) using branch lengths and model parameters resulting from the constrained ML analysis. Each simulated data set was then analyzed under ML twice: once under topological constraints to enforce monophyly of the clade of interest and once with no topological constraints. The distribution for the test statistic was generated by calculating the difference in lnL scores between the best unconstrained tree and the best constrained tree for each simulated data set. The lnL difference between the best constrained and unconstrained trees for the original data set was then directly compared to the appropriate distribution to estimate p values.
Results
Sequence Comparisons and Practical Considerations
Evaluation of sequences collected in this study supports the view that they represent coding region sequences of the ATPase α-subunit gene. Initial comparisons of these sequences with ATPase sequences in GenBank using the BLAST search protocol returned all ATPase α-subunit coding sequences as top matches. There is evidence of among-sequence base composition heterogeneity when all sequences and all codon positions are evaluated (mean nucleotide frequencies: A = 0.25113, C = 0.24410, G = 0.25153, and T = 0.25324 [p ‹ 0.01, χ2 test of homogeneity of base frequencies across taxa]), but the heterogeneity appears to be confined to third codon positions. With third positions excluded, the mean nucleotide frequencies are A = 0.29011, C = 0.20925, G = 0.25809, and T = 0.24255, and there is no significant heterogeneity (p = 0.94098848).
Unfortunately, the primers developed here are not universal metazoan primers—despite several attempts, the primers failed to amplify the target fragment from RNA extracts from two bryozoans (Bugula nerita and Pectinatella magnifica), two nematodes (Camallanus oxycephalus and an unidentified mermithid nematode), a nematomorph (Gordius sp.), two sipunculans (Golfingia pugettensis and Phascolosoma agassizii), an echiuran (Urechiscaupo), two echinoderms (the holothuroid Eupentactaquinquesemita and the ophiuroid Ophiothrix spiculata), an apterygote insect (Thermobia domestica), and four annelids (the polychaetes Nereis virens and Phragmatopoma californica, the hirudinid Placobdella parasitica, and an unidentified lumbricid earthworm). For two of the three gastropods studied (Tegula brunnea and Onchidella borealis), only about 1000 bp could be successfully amplified and sequenced. Entire sequences were obtained for the pseudoscorpion Garypus californica and the brown recluse spider Loxosceles reclusa, but some regions had several ambiguous base calls. The practical value of this gene region for studies of bilaterian phylogeny is admittedly somewhat limited if universal PCR primers cannot be developed. Development of consensus-degenerate hybrid oligonucleotide (a.k.a. CODEHOP) primers (Rose et al. 1998), which employ a degenerate “core” region at the 3′ end of the primer and a nondegenerate consensus “clamp” region at the 5′ end, may allow amplification of this gene fragment from additional taxa; we are currently exploring this possibility.
Phylogenetic Analyses
Nucleotide Data
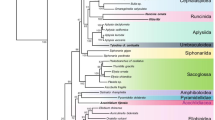
The nucleotide and amino acid data matrices, as well as several trees, have been submitted to TreeBASE (www.treebase.org ; study accession number = S904, matrix accession numbers = M1485–M1487). MP analysis of the small nucleotide alignment (including only isoform 1 sequences from vertebrates) yielded 32 equally parsimonious trees (TL = 3819; trees not shown). Successive weighting and implicit weight MP analyses, as well as ME analysis of LogDet + I distances, each yielded one tree (not shown). ML analysis yielded a single tree (Figs. 1 and 2; InL = 19,806.34902162). Several clades are recovered with high bootstrap support and Bayesian clade posterior probability. A large clade comprising all mollusk, nemertean, flatworm, and bryozoan sequences, along with the phoronid and brachiopod sequences, was recovered with moderate support (ML bootstrap support value [LBS] = 77, MP bootstrap support value [PBS] = 62, Bayesian posterior probability [BPP] = 1.0). This clade, known as Lophotrochozoa (Halanych et al. 1995) or Eutrochozoa (Eernisse et al. 1992; Ghiselin 1989), has been recovered in one form or another in several recent studies (e.g., Peterson and Eernisse 2001). Within this group, only a bryozoan clade (LBS and PBS = 100, BPP = 1.0), a mollusk clade (LBS = 65, PBS = 63, BPP = 0.986), and a pairing of the two bivalve sequences (LBS = 84, PBS = 89, BPP = 1.0) are supported. It should be noted, however, that taxon sampling is extremely limited and that future studies of this gene that include more taxa may reach different conclusions.

Maximum likelihood tree for the small taxon set (including only vertebrate isoform I sequences), with groups of interest highlighted. Support values are as follows: above branches (left)—equal-weights parsimony bootstrap support values; above branches (right)—maximum likelihood bootstrap support values; below branches—Bayesian clade posterior probabilities. Branches marked with a single boldface italicized “100” had 100% MP and ML bootstrap support. An asterisk (*) indicates a partial sequence (approximately 1000 bp). Letters denote the following clades of interest: A—Echinodermata, B—Bryozoa (Cheilostomata), C—Platyhelminthes, D—Bivalvia, E—Gastropoda, F—Myriapoda, G—Arachnida, H—Coleoptera, I—Diptera and J—“Dermaptera–Hemiptera–Siphonoptera” (DHS) clade.

Maximum likelihood phylogram for the small taxon set. The scale bar is equivalent to 0.05 substitution per site.
Another large clade, consisting of the lone nematode sequence and all arthropod sequences, was also recovered with moderate support (LBS = 61, PBS = 49, BPP = 1.0), although the position of the nematode sequence (as sister to Artemia, a brine shrimp; LBS = 72, PBS = 49, BPP = 1.0) is unexpected. The LogDet + Invariant ME tree has the Caenorhabditis sequence in a different position—as sister to a Crustacea + Hexapoda clade—but Arthropoda itself is still not monophyletic (Table 3). Within Arthropoda, arachnid (LBS = 97, PBS = 97, BPP = 1.0), hexapod (LBS/PBS = 100, BPP = 1.0), and myriapod (LBS = 81, PBS = 63, BPP = 0.991) monophyly are well supported. An opilionid + pseudoscorpion grouping (LBS = 97, PBS = 89, BPP = 1.0) and monophyly for a group of malacostracan crustaceans consisting of an isopod, an amphipod, and two decapods (LBS/PBS = 100, BPP = 1.0) are also strongly supported. Hexapods are found to be nested within Crustacea (including the Artemia + Caenorhabditis clade). Within Hexapoda, dipteran monophyly (LBS = 96, PBS = 89, BPP = 1.0), coleopteran monophyly (LBS= 82, PBS = 64, BPP = 0.999) and monophyly for a dermapteran + hemipteran + siphonopteran clade (LBS = 96, PBS = 83, BPP = 1.0) are all moderately supported.
A monophyletic Deuterostomia is not recovered in these analyses (i.e., the echinoderms do not form a clade with the vertebrates; Fig. 1). There is, however, appreciable support for a pairing of the two echinoderms (LBS = 77, PBS = 71, BPP = 1.0). Vertebrate monophyly is also strongly supported (LBS/PBS = 100, BPP = 1.0), as is monophyly of Tetrapoda (LBS = 91, PBS = 91, BPP = 1.0), Amniota (LBS = 95, PBS = 90, BPP = 1.0), and Mammalia (LBS/PBS = 100, BPP = 1.0), although relationships within Mammalia are poorly supported.
All MP and ME trees, as well as the majority-rule consensus tree from Bayesian analysis, are highly congruent with the ML tree (Table 3). The major differences between trees resulting from different analyses are in regions of low bootstrap support and Bayesian posterior probability, particularly within Lophotrochozoa and Arthropoda. Deuterostome monophyly is not recovered in any analysis of the small nucleotide alignment, and some analyses do not recover a Myriapoda + Arachnida clade or a monophyletic Myriapoda (Table 3). Monophyly for Gastropoda, Coleoptera, Osteicththyes, Echinodermata, Protostomia, the Artemia + Caenorhabditis clade, and the “Diptera + DHS” clade are also recovered in most, but not all, analyses.
MP analysis of the large nucleotide alignment (including isoform 1, 2, 3, and 4 sequences from several vertebrates) yielded eight equally parsimonious trees (TL = 4679; trees not shown). As above, successive weighting and implicit weighting MP analyses produced one tree, as did ME analysis of LogDet + I distances (not shown). ML analysis yielded a single tree (Fig. 3; InL = 23871.83723707) that is congruent with the ML tree for the small taxon set (Fig. 1) in all but one respect. ML analysis of the large taxon set recovered a monophyletic Osteichthyes (all vertebrates but Torpedo californica, a cartilaginous fish) in the isoform 1 clade with low support (LBS = 52, PBS = 45, BPP = 0.642). A monophyletic Osteichthyes was not recovered in ML analysis of the small taxon set (Fig. 1).

Maximum likelihood tree for the large taxon set with different isoform groups highlighted. Support values are as described for Fig. 1.
Amino Acid Data
Equal-weights MP analysis of the amino acid alignment resulted in six trees (TL = 2743; strict consensus shown in Fig. 4). Implicit weights parsimony and successive weighting analyses each produced one tree (not shown). The amino acid parsimony trees are highly congruent with trees recovered in analyses of the nucleotide data (Table 3). The only major difference between the amino acid trees and the nucleotide trees is the lack of support for a Myriapoda + Arachnida clade and variable recovery of a monophyletic Myriapoda in the amino acid trees (Table 3). The quartet puzzling ML tree for the amino acid data (not shown) is less resolved than—but largely congruent with—the amino acid MP trees and nucleotide trees, with a notable exception; Pantinonemertes is not found with the flatworms (as for DNA data) but as sister group to Lineus.

The tree resulting from equal-weights maximum parsimony analysis of the amino acid data matrix. Values above the branches are maximum parsimony bootstrap support values, and values below the branches are maximum likelihood quartet puzzling support values (WAGf + Γ + I model); only values above 50 are shown.
Parametric Bootstrapping Analyses
The results of the parametric bootstrapping tests of deuterostome, arthropod, and nemertean monophyly are shown in Fig. 5. Deuterostome monophyly cannot be rejected by these data (1.0 > p > 0.05), while arthropod and nemertean monophyly are rejected (p < 0.01 in both cases).

Results of the parametric bootstrapping analyses. The −lnL difference between the best constrained and the best unconstrained trees for each test is denoted by an arrow. a Test of monophyly for Deuterostomia (including 2 echinoderm and 13 vertebrate sequences). b Test of monophyly for Arthropoda (19 arthropod sequences). c Test of monophyly for Nemertea (three nemertean sequences). The difference in likelihood scores for the original data were 1.10542415 (Deuterostomia monophyly;1.0 > p > 0.05), 20.44706 (Arthropoda monophyly; p < 0.01, and 12.90524706 (Nemertea monophyly; p < 0.01). Please see the text for further details.
Discussion
Phylogenetic Hypotheses
In general, phylogenetic analyses of this fragment of the sodium–potassium ATPase α-subunit result in trees that are concordant with results of previous studies of metazoan phylogeny based on morphological and/or 18S sequence data. Monophyly of most phyla is supported by the ATPase analyses, as is monophyly of a few major superphylum-level groups (Protostomia, Ecdysozoa, and Lophotrochozoa).
Relationships Within Protostomia
Recovery of a variant of Ecdysozoa—a clade consisting of all molting animals sampled for this study—is consistent with previous findings based on both 18S data (Aguinaldo et al. 1997) and some analyses of morphological data (Eernisse et al. 1992). Although a monophyletic Ecdysozoa was recovered in all analyses, the position of the C. elegans sequence—usually as sister to Artemia franciscana— was unexpected (this finding is evaluated further below). Overall, most other inferred relationships within Arthropoda match previous findings rather closely. The presence of a Pancrustacea clade (disregarding the enigmatic position of the C. elegans sequence) in the ATPase phylogeny serves as independent support of this finding in studies of other sources of data, including 18S (Giribet and Ribera 2000), EF-1α (Regier and Shultz 1997), combined morphology and 18S (Zrzavy et al. 1998), a combined data set consisting of several genes and morphology (Giribet et al. 2001), and mitochondrial gene order (Boore et al. 1998). The positions of Myriapoda and Chelicerata remain unclear, with some previous studies proposing a Myriapoda + Chelicerata clade (Friedrich and Tautz 1995; Giribet et al. 1996; Hwang et al. 2001; Kusche and Burmester 2001; Turbeville et al. 1991), while others support a basal position for Myriapoda (Regier and Shultz 2001) or Chelicerata (Regier and Shultz 1997; Shultz and Regier 2000; Wheeler et al. 1993) within Arthropoda. A Myriapoda + Chelicerata clade is supported by a putative molecular synapomorphy—a deletion of the second α-helix of hemocyanin domain 1 (Kusche and Burmester 2001). Support for any particular resolution of myriapod/chelicerate relationships is, however, generally low in most studies. The ATPase nucleotide data favor a Myriapoda + Chelicerata (or, more accurately, Arachnida) pairing, although bootstrap support and posterior probability are not high (Fig. 1). Analyses of the amino acid data set, however, do not support a Myriapoda + Chelicerata clade (Fig. 4). Within Arachnida, the ATPase data generally support a close relationship between an opilionid (harvestman) and Garypus californica (a pseudoscorpion), with Loxosceles reclusa (a spider) basal to this pair. Although relationships within Arachnida remain unclear, this finding is also not unprecedented (see, e.g., Figs. 3,4,5 of Giribet and Ribera 2000).
Within Pancrustacea, a pairing of two brachyuran crabs (Carcinus and Callinectes) and monophyly for Malacostraca (represented here by a caprellid amphipod, a terrestrial isopod, and the two brachyurans) are unsurprising. The ATPase data suggest, however, that Crustacea is paraphyletic not only with respect to Hexapoda, but also with respect to Nematoda, a rather unlikely proposition. The only nonmalacostracan crustacean sequence in the analysis (Artemia franciscana, a branchiopod) lies closer to hexapods than to the malacostracan sequences. This finding is also incongruent with some recent publications based on 18S sequences (Giribet and Wheeler 1999) and complete mitochondrial genome sequences (Wilson et al. 2000), which propose a close relationship between malacostracans and hexapods, with branchiopods in a relatively basal position. Within Hexapoda, a monophyletic Coleoptera and Diptera were expected. However, the “DHS” (Dermaptera–Hemiptera–Siphonoptera) clade, and the hemipteran–flea pairing found within this clade, is radically different from previous hypotheses of hexapod relationships (Wheeler et al. 2001 and citations therein). With respect to the hexapod taxa included here, Wheeler et al, (2001) proposed a relatively close relationship between Siphonoptera and Diptera, with Hemiptera and Dermaptera outside this pairing in successively basal positions, a hypothesis clearly different from the ATPase tree. Taxon sampling is unquestionably an issue here—Wheeler and coworkers’ study included 128 terminal taxa representing 33 hexapod orders, while only 8 sequences from 5 hexapod orders are included here.
Within Lophotrochozoa, apart from some phylum-level groupings (e.g., Bryozoa and Mollusca) and a phoronid–brachiopod pair, there is very little support for any particular set of interphylum relationships. Unfortunately, this has been the case in most molecular phylogenetic studies of relationships within this clade (e.g., Mallatt and Winchell 2002; Winnepenninckx et al. 1995). The majority of clades within Lophotrochozoa are supported in fewer than 50% of ML and MP bootstrap replicates, and Bayesian clade posterior probabilities are also relatively low (below 0.9) for several groups. It is becoming increasingly clear that single molecules (18S, EF-1α and ATPase) are insufficient to resolve interphylum relationships within Lophotrochozoa. Recently, several researchers have turned to analyses of combined data (both morphological and molecular) to resolve these issues (Mallatt and Winchell 2002; Zrzavy et al. 1998), thus far with limited success. It appears, however, that only combined analyses of multiple data sets will resolve relationships within this problematic group.
Two clades that are recovered with relatively high support on the ATPase ML tree are Mollusca and, within Mollusca, Bivalvia. In contrast, most studies of 18S data alone have failed to resolve a monophyletic Mollusca or, for that matter, a monophyletic Bivalvia (Adamkewicz et al. 1997; Canapa et al. 1999; Steiner and Muller 1996; Winnepenninckx et al. 1996). Taxon sampling within Mollusca in this study is very limited, however, and with additional molluscan exemplars, ATPase may also reveal disconcerting patterns of molluscan para- or polyphyly. This is particularly true for Bivalvia, as the two bivalve species sampled here—Lampsilis cardium (a freshwater unionid) and Macoma nasuta (a marine tellinid)—are fairly closely related (Brusca and Brusca 1990). More extensive taxon sampling is clearly necessary before strong conclusions can be made regarding mollusk (or bivalve) monophyly and the utility of this gene for resolving relationships within Mollusca or Lophotrochozoa.
Relationships Within Deuterostomia
Apart from the peculiar rooting of the tree between the echinoderms and the vertebrates (discussed further below), this gene fragment successfully recovered nearly all postulated relationships within Deuterostomia, generally with high clade posterior probabilities and MP and ML bootstrap support. Mammalia (represented here by Rattus norvegicus, Ovis aries, Sus scrofa, Canis familiaris, and Homo sapiens), Amniota (Gallus gallus + Mammalia), Anura (Bufo marinus + Xenopus laevis), Tetrapoda (Anura + Amniota), and Actinopterygii (Anguilla anguilla, Catostomus commersoni, Danio rerio, and Oreochromis mossambicus) are all strongly supported, and Osteichthyes (Actinopterygii + Sarcopterygii, represented here only by various tetrapods) is also recovered in ML analyses of the large data set (Fig. 3). However, neither tree is completely concordant with traditional views of relationships within Actinopterygii or Mammalia. Within Actinopterygii, Anguilla (a relatively basal actinopterygian) is expected to be sister to the other three actinopterygian species (Lauder and Liem 1983), although support for a C. commersoni–D. rerio clade was expected (as both species are cypriniforms). Support for branches within this region of the tree is not high, so this may simply be a region of the tree that ATPase sequences are unable to resolve accurately. Support for any particular hypothesis of relationships within Mammalia is also low, as has been seen in many other studies of mammal ordinal relationships (see Waddell et al. 1999b and citations therein).
Parametric Bootstrapping Tests
Despite the general congruence between the ATPase phylogeny and previous findings, some aspects of the trees presented here were surprising. Three of the most peculiar findings—deuterostome, arthropod, and nemertean para/polyphyly—were investigated using parametric bootstrapping tests. There is substantial molecular and morphological support for monophyly of these groups (particularly Arthropoda and Deuterostomia), yet nearly all analyses of the ATPase fragment fail to recover these groups. The parametric bootstrapping tests of deuterostome, arthropod, and nemertean monophyly produced mixed results—deuterstome monophyly could not be rejected, but nemertean and arthropod monophyly was rejected. If the position of the outgroup Hydra sequence is ignored, the ATPase data do support deuterostome monophyly. The difference in likelihood scores between a “deuterostome monophyly” tree and the best ML topology is insignificant (p > 0.08; Fig. 5). The branch leading to the Hydra sequence is connected to the longest internal branch on the ML tree (the branch between the basal vertebrate node and the rest of the tree; Fig. 2), suggesting that long-branch attraction—even weak attraction, given the lack of signal supporting Deuterostomia—may be playing a role. Removal of the Hydra sequence prior to ML analysis produces a nearly identical topology, but on this unrooted phylogeny the two echinoderm sequences are no longer sisters—yielding the topology ((((Vertebrata) Strongylocentrotus) Asterina) all other sequences)—and the Phoronopsis + Terebratalia clade is sister to the Hubrechtella + Lineus clade (tree not shown). This loss of echinoderm monophyly when the Hydra sequence is excluded suggests that inclusion of even this single outgroup sequence may aid resolution in parts of the phylogeny. Additional outgroup sequences (from ctenophores, other cnidarians, or sponges) or sequences from other deuterostomes (e.g., hemichordates, urochordates, or cephalochordates) may allow a monophyletic Deuterostomia to be recovered. Some analyses of the alignment including vertebrate ATPase isoforms 2, 3, and 4 do recover deuterostome monophyly (trees not shown), suggesting that more thorough taxon sampling may alleviate this problem.
Parametric bootstrapping analysis rejects arthropod monophyly (p < 0.01; Fig. 5), but the rejection is solely due to the nematode (C. elegans) sequence. The nematode branch is one of the longest branches on the ML tree, and in this case, it is joined to the second-longest terminal branch (leading to Artemia) (Fig. 2). The parametric bootstrapping analysis strongly rejects arthropod monophyly, however, and sequential exclusion of the Caenorhabditis and Artemia sequences from the data set produces ML trees that are identical to those produced when both sequences are included (trees not shown). Sodium–potassium ATPase is known to play a critical role in the extraordinary osmoregulatory abilities of brine shrimp. The Na+/K+ α-subunit gene in Artemia has been studied extensively by Saez et al. (2000), who found higher levels of polymorphism in Na+/K+ ATPase α-subunit isoform 1 sequences than expected and suggested that this locus is evolving under positive selection. This could explain the high relative rate of change of this gene in Artemia and the seemingly spurious grouping of the Artemia sequence with the Caenorhabditis sequence.
Nemertean monophyly, based on the three sequences included here, is rejected by parametric bootstrapping analysis (p < 0.01; Fig. 5) and is also not recovered in any analysis of either the nucleotide or the amino acid data. This result can be examined in light of a recent phylogenetic study of nemertean relationships. Based on analyses of 16S, 28S, histone H3, and COI gene regions, Thollesson and Norenburg (2003) have proposed that Nemertea comprises two major clades—Hoplonemertea and Pilidiophora (a group comprising Heteronemertea and Hubrechtella)—and a paraphyletic “paleonemertean” assemblage. Two of the nemertean sequences included here are members of Pilidiophora (Lineus and Hubrechtella), while the other is a member of Hoplonemertea (Pantinonemertes sp. nov.). A Lineus + Hubrechtella pair is recovered in nearly all analyses based on DNA sequences, but the Pantinonemertes sequence typically forms a clade with the two platyhelminth sequences (from Dugesia and Schistosoma). In the quartet puzzling ML analysis of amino acid sequences, however, Pantinonemertes and Lineus form a clade to the exclusion of Hubrechtella, and there is no support for a nonmonophyletic Nemertea (the position of Hubrechtella is unresolved). It is thus likely that the odd result is an artifact in the analytical methods rather than a case of paralogy or sample contamination. As with other genes that have been used to evaluate metazoan relationships (e.g., 18S), there is low support for most clades within Lophotrochozoa on the ATPase trees (Figs. 1 and 4), but the peculiar position of Pantinonemertes (and the rejection of nemertean monophyly in the parametric bootstrapping test) merits further exploration.
The Phylogenetic Utility of the Sodium–Potassium ATPase α-Subunit Gene
Will the sodium–potassium ATPase α-subunit gene region studied here be useful for future deep-level studies of animal phylogeny? Practical as well as empirical considerations are required to address this question fully. To be of practical use for molecular phylogenetic studies, the fragment must be amenable to PCR amplification across several phyla (or at least within phyla of interest). The gene should also be single copy, so that erroneous comparisons among paralogous gene copies can be avoided (Martin and Burg 2002). Alternatively, if there are multiple copies of the gene, sequences in the PCR priming region should allow PCR to amplify only orthologs. Empirical considerations might include the ability of a novel gene fragment to recover clades that are well supported based on studies of morphological data or sequence data from other genes. In addition, the ability of a new phylogenetic marker to resolve relationships that other data sets have failed to resolve (or to aid in the resolution of such relationships in combination with other sources of data) may also suggest that a new marker can provide an important contribution.
Is the ATPase fragment studied here practically useful for metazoan phylogenetic studies? We believe that it can be useful for some questions. The gene was successfully amplified from representatives of seven phyla (Arthropoda, Brachiopoda, Bryozoa, Echinodermata, Mollusca, Phoronida, and Nemertea) and is particularly easy to amplify from some subgroups (e.g., Hexapoda). Future studies of relationships within groups where amplification was least problematic may benefit from inclusion of data from this gene. Unfortunately, the failure of PCR amplification for representatives of several phyla (most notably annelids, sipunculans, cnidarians, and nematodes) limits the usefulness of these primer pairs in studies of relationships among the metazoan phyla. Future work on this gene will involve further attempts to amplify the gene from additional phyla and evaluation of additional primer pairs designed using the CODEHOP approach described above.
There is some evidence from previous studies (Baxter-Lowe et al. 1989; Friedlander et al. 1992) that this gene is multicopy in some taxa. In this study, several ambiguities were found in two of the three arachnids sequenced (Garypus and Loxosceles), suggesting that there may be two (or more) functional copies of this gene in at least some arachnids. The recovery of a monophyletic Arachnida suggests that this putative duplication may only be an issue within this group. Any future phylogenetic studies using this gene will require a more thorough examination of gene copy number in the taxon (or taxa) of interest.
Finally, are there empirical benefits to the use of this ATPase α-subunit gene region in metazoan phylogenetic studies? The divergences between taxa studied here span a broad range of time. For example, minimum estimated time depths based on other data for splits among the major vertebrate groups represented here range from 65/92 million years ago (mya) for the split between carnivores and primates all the way to 439/528 mya (the split between Chondrichthyes and Osteicthyes). Between these extremes lie the synapsid–diapsid split (310 mya), the Lissamphibia–Amniota split (323/360 mya), and the Actinopterygii–Sarcopterygia split (424/450 mya) (all pairs of dates are crude fossil-based estimates derived from several sources [Alroy 1999; Benton 1997; Janvier 1996; Zug et al. 2001] and molecular clock-based estimates [Kumar and Hedges 1998; Waddell et al. 1999a], respectively). The finding that the Na+/K+ ATPase α-subunit accurately recovers these splits with high support suggests that splits of similar time depth within other metazoan phyla might be investigated fruitfully using this gene. In addition, the Lophotrochozoa/Ecdysozoa split occurred at least 560 mya, given fossil considerations and a recent Bayesian estimate of the minimum divergence time for these two clades (Aris-Brosou and Yang 2002). The recovery of these two clades in all analyses, along with generally reasonable patterns of relationships within Vertebrata, suggests that this gene fragment may be useful for both intra- and interphylum relationships within Metazoa, particularly in combination with data from other genes.
References
SL Adamkewicz MG Harasewych J Blake D Saudek CJ Bult (1997) ArticleTitleA molecular phylogeny of the bivalve mollusks. Mol Biol Evol 14 619–629 Occurrence Handle1:STN:280:ByiA3szpsVU%3D Occurrence Handle9190063
AMA Aguinaldo JM Turbeville LS Lindford MC Rivera JR Garey RA Raff JA Lake (1997) ArticleTitleEvidence for a clade of nematodes, arthropods and other moulting animals. Nature 387 389–393 Occurrence Handle10.1038/387489a0
J Alroy (1999) ArticleTitleThe fossil record of North American mammals: Evidence for a Paleocene evolutionary radiation. Syst Biol 48 107–118 Occurrence Handle10.1080/106351599260472 Occurrence Handle1:STN:280:DC%2BD38zjslSjtg%3D%3D Occurrence Handle12078635
FE Anderson (2000) ArticleTitlePhylogeny and historical biogeography of the loliginid squids (Mollusca: Cephalopoda) based on mitochondrial DNA sequence data. Mol Phylogenet Evol 15 191–214 Occurrence Handle10.1006/mpev.1999.0753 Occurrence Handle1:CAS:528:DC%2BD3cXjs12qtbg%3D Occurrence Handle10837151
S Aris-Brosou ZH Yang (2002) ArticleTitleEffects of models of rate evolution on estimation of divergence dates with special reference to the metazoan 18S ribosomal RNA phylogeny. Syst Biol 51 703–714 Occurrence Handle10.1080/10635150290102375 Occurrence Handle12396585
LA Baxter-Lowe JZ Guo EE Bergstrom LE Hokin (1989) ArticleTitleMolecular cloning of the Na,K-ATPase alpha-subunit in developing brine shrimp and sequence comparison with higher organisms. FEBS Lett 257 181–187 Occurrence Handle10.1016/0014-5793(89)81816-2 Occurrence Handle1:CAS:528:DyaK3cXitlSmu78%3D Occurrence Handle2553490
MJ Benton (1997) Vertebrate palaeontology. Chapman & Hall London
JE Blair K Ikeo T Gojobori SB Hedges (2002) ArticleTitleThe evolutionary position of nematodes. BMC Evol Biol 2 7 Occurrence Handle10.1186/1471-2148-2-7 Occurrence Handle11985779
M Blanchette T Kunisawa D Sankoff (1999) ArticleTitleGene order breakpoint evidence and animal mitochondrial phylogeny. J Mol Evol 49 193–203 Occurrence Handle1:CAS:528:DyaK1MXltFSktrw%3D Occurrence Handle10441671
JL Boore (1999) ArticleTitleAnimal mitochondrial genomes. Nucleic Acids Res 27 1767–1780 Occurrence Handle10.1093/nar/27.8.1767 Occurrence Handle1:CAS:528:DyaK1MXivVersbo%3D Occurrence Handle10101183
JL Boore WM Brown (2000) ArticleTitleMitochondrial genomes of Galathealinum, Helobdella, and Platynereis: Sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol 17 87–106 Occurrence Handle1:CAS:528:DC%2BD3cXot1Sjsw%3D%3D Occurrence Handle10666709
JL Boore JL Staton (2002) ArticleTitleThe mitochondrial genome of the sipunculid Phascolopsis gouldii supports its association with Annelida rather than Mollusca. Mol Biol Evol 19 127–137 Occurrence Handle1:CAS:528:DC%2BD38XovVCjtg%3D%3D Occurrence Handle11801741
JL Boore DV Lavrov WM Brown (1998) ArticleTitleGene translocation links insects and crustaceans. Nature 392 667–668 Occurrence Handle10.1038/33577 Occurrence Handle1:CAS:528:DyaK1cXjtVWmuro%3D Occurrence Handle9565028
C Borchiellini N Boury-Esnault J Vacelet Y Le Parco (1998) ArticleTitlePhylogenetic analysis of the Hsp70 sequences reveals the monophyly of Metazoa and specific phylogenetic relationships between animals and fungi. Mol Biol Evol 15 647–655 Occurrence Handle1:CAS:528:DyaK1cXjsFShsbc%3D Occurrence Handle9615446
RC Brusca GJ Brusca (1990) Invertebrates. Sinauer Associates Sunderland, MA
A Canapa I Marota F Rollo E Olmo (1999) ArticleTitleThe small-subunit rRNA gene sequences of venerids and the phylogeny of Bivalvia. J Mol Evol 48 463–468 Occurrence Handle1:CAS:528:DyaK1MXhvFyqu7s%3D Occurrence Handle10079284
Y Cao PJ Waddell N Okada M Hasegawa (1998) ArticleTitleThe complete mitochondrial DNA sequence of the shark Mustelus manazo: Evaluating rooting contradictions to living bony vertebrates. Mol Biol Evol 15 1637–1646 Occurrence Handle1:CAS:528:DyaK1MXjtVOl Occurrence Handle9866199
J Castresana G Feldmaier-Fuchs S Yokobori N Satoh S Paabo (1998) ArticleTitleThe mitochondrial genome of the hemichordate Balanoglossus carnosus and the evolution of deuterostome mitochondria. Genetics 150 1115–1123 Occurrence Handle1:CAS:528:DyaK1cXnsFCntLo%3D Occurrence Handle9799263
S Cho A Mitchell JC Regier C Mitter RW Poole TP Friedlander S Zhao (1995) ArticleTitleA highly conserved nuclear gene for low-level phylogenetics: Elongation factor-1 alpha recovers morphology-based tree for heliothine moths. Mol Biol Evol 12 650–656 Occurrence Handle1:CAS:528:DyaK2MXmsVOqs7c%3D Occurrence Handle7659020
R Christen A Ratto A Baroin R Perasso KG Grell A Adoutte (1991) ArticleTitleAn analysis of the origin of metazoans, using comparisons of partial sequences of the 28S RNA, reveals an early emergence of triploblasts. EMBO J 10 499–503 Occurrence Handle1:CAS:528:DyaK3MXhvVSmsbo%3D Occurrence Handle2001670
R de Rosa JK Grenier T Andreeva CE Cook A Adoutte M Akam SB Carroll G Balavoine (1999) ArticleTitleHox genes in brachiopods and priapulids and protostome evolution. Nature 399 772–776 Occurrence Handle10.1038/21631 Occurrence Handle1:STN:280:DyaK1MzhvVWhtw%3D%3D Occurrence Handle10391241
M Dowtin AD Austin (1999) ArticleTitleEvolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol 16 298–309 Occurrence Handle10028295
I Eeckhaut D McHugh P Mardulyn R Tiedemann D Monteyne M Jangoux MC Milinkovitch (2000) ArticleTitleMyzostomida: A link between trochozoans and flatworms? Proc R Soc Lond B 267 1383–1392 Occurrence Handle10.1098/rspb.2000.1154 Occurrence Handle1:STN:280:DC%2BD3M7gsV2htg%3D%3D
DJ Eernisse JS Albert FE Anderson (1992) ArticleTitleAnnelida and Arthropoda are not sister taxa: A phylogenetic analysis of spiralian metazoan morphology. Syst Biol 41 305–330
A Erber D Riemer M Bovenschulte K Weber (1998) ArticleTitleMolecular phylogeny of metazoan intermediate filament proteins. J Mol Evol 47 751–762 Occurrence Handle1:CAS:528:DyaK1cXotVGhsr4%3D Occurrence Handle9847417
A Erber D Riemer H Hofemeister M Bovenshulte R Stick G Panopoulou H Lebrach K Weber (1999) ArticleTitleCharacterization of the Hydra lamin and its gene: A molecular phylogeny of metazoan lamins. J Mol Evol 49 260–271 Occurrence Handle1:CAS:528:DyaK1MXltFSrtb0%3D Occurrence Handle10441677
J Felsenstein (1985) ArticleTitleConfidence limits on phylogenies: An approach using the bootstrap. Evolution 39 783–791
J Felsenstein (1995) PHYLIP (phylogeny inference package). Distributed by the author, Department of Genetics, University of Washington Seattle
KG Field GJ Olsen DJ Lane SJ Giovannoni MT Ghiselin EC Raff NR Pace RA Raff (1988) ArticleTitleMolecular phylogeny of the animal kingdom. Science 239 748–753 Occurrence Handle1:CAS:528:DyaL1cXhsVKltb8%3D Occurrence Handle3277277
P Flook H Rowell G Gellissen (1995) ArticleTitleHomoplastic rearrangements of insect mitochondrial transfer-RNA genes. Naturwissenschaften 82 336–337 Occurrence Handle1:CAS:528:DyaK2MXntFKit7w%3D
F Frati C Simon J Sullivan DL Swofford (1997) ArticleTitleEvolution of the mitochondrial cytochrome oxidase II gene in Collembola. J Mol Evol 44 145–158 Occurrence Handle1:CAS:528:DyaK2sXhtFaksb0%3D Occurrence Handle9069175
TP Friedlander JC Regier C Mitter (1992) ArticleTitleNuclear gene sequences for higher level phylogenetic analysis: 14 promising candidates. Syst Biol 41 483–489
M Friedrich D Tautz (1995) ArticleTitleRibosomal DNA phylogeny of the major extant arthropod classes and the evolution of myriapods. Nature 376 165–167 Occurrence Handle10.1038/376165a0 Occurrence Handle1:CAS:528:DyaK2MXmvFalsbs%3D Occurrence Handle7603566
MT Ghiselin (1989) Summary of our present knowledge of metazoan phylogeny. B Fernholm K Bremer H Jörnvall (Eds) The hierarchy of life. Elsevier Amsterdam 261–272
G Giribet C Ribera (1998) ArticleTitleThe position of arthropods in the animal kingdom: A search for a reliable outgroup for internal arthropod phylogeny. Mol Phylogenet Evol 9 481–488 Occurrence Handle10.1006/mpev.1998.0494 Occurrence Handle1:STN:280:DyaK1czjsVGksw%3D%3D Occurrence Handle9667996
G Giribet C Ribera (2000) ArticleTitleA review of arthropod phylogeny: New data based on ribosomal DNA sequences and direct character optimization. Cladistics 16 204–231 Occurrence Handle10.1006/clad.1999.0128
G Giribet WC Wheeler (1999) ArticleTitleThe position of arthropods in the animal kingdom: Ecdysozoa, islands, trees and the “parsimony ratchet. Mol Phylogenet Evol 13 619–623 Occurrence Handle10.1006/mpev.1999.0679 Occurrence Handle1:STN:280:DC%2BD3c%2FovVOisg%3D%3D Occurrence Handle10620418
G Giribet J Carranza J Baguñà M Riutort C Ribera (1996) ArticleTitleFirst molecular evidence for the existence of a Tardigrada + Arthropoda clade. Mol Biol Evol 13 76–84 Occurrence Handle1:CAS:528:DyaK28XhtVylur8%3D Occurrence Handle8583909
G Giribet DL Distel M Polz W Sterrer WC Wheeler (2000) ArticleTitleTriploblastic relationships with emphasis on the acoelomates and the position of Gnathostomulida, Cycliophora, Plathelminthes, and Chaetognatha: A combined approach of 18S rDNA sequences and morphology. Syst Biol 49 539–562 Occurrence Handle1:STN:280:DC%2BD38zntVKquw%3D%3D Occurrence Handle12116426
G Giribet GD Edgecombe WC Wheeler (2001) ArticleTitleArthropod phylogeny based on eight molecular loci and morphology. Nature 413 157–161 Occurrence Handle10.1038/35093097 Occurrence Handle1:CAS:528:DC%2BD3MXntVOqsbY%3D Occurrence Handle11557979
KM Halanych (1998) ArticleTitleConsiderations for reconstructing metazoan history: Signal, resolution, and hypothesis testing. Am Zool 38 929–941
KM Halanych JD Bacheller AM Aguinaldo SM Liva DM Hillis JA Lake (1995) ArticleTitleEvidence from 18S ribosomal DNA that the lophophorates are protostome animals. Science 267 1641–1643 Occurrence Handle1:CAS:528:DyaK2MXksVajtbk%3D Occurrence Handle7886451
MJ Hickerson CW Cunningham (2000) ArticleTitleDramatic mitochondrial gene rearrangements in the hermit crab Pagurus longicarpus (Crustacea, Anomura). Mol Biol Evol 17 639–644 Occurrence Handle1:CAS:528:DC%2BD3cXisVSgtLo%3D Occurrence Handle10742054
DM Hillis B Mable C Moritz (1996) Applications of molecular systematics and the future of the field. DM Hillis B Mable C Moritz (Eds) Molecular systematics. Sinauer Associates Sunderland, MA 515–543
JP Huelsenbeck DM Hillis R Jones (1996) Parametric bootstrapping in molecular phylogenetics: Applications and performance. JD Ferraris SR Palumbi (Eds) Molecular zoology: Advances, strategies and protocols. Wiley–Liss New York 19–46
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogeny. http://morphbank.ebc.uu.se/mrbayes/
UW Hwang M Friedrich D Tautz CJ Park W Kim (2001) ArticleTitleMitochondrial protein phylogeny joins myriapods with chelicerates. Nature 413 154–157 Occurrence Handle1:CAS:528:DC%2BD3MXntVOqsLc%3D Occurrence Handle11557978
P Janvier (1996) Early vertebrates. Clarendon Press New York
M Kobayashi H Wada N Satoh (1996) ArticleTitleEarly evolution of the Metazoa and phylogenetic status of the diploblasts as inferred from amino acid sequence of elongation factor-1α. . Mol Phylogenet Evol 5 414–422 Occurrence Handle1:CAS:528:DyaK28XjtVCku7s%3D Occurrence Handle8728399
S Kojima (1998) ArticleTitleParaphyletic status of Polychaeta suggested by phylogenetic analysis based on the amino acid sequences of elongation factor-1α. Mol Phylogenet Evol 9 255–261 Occurrence Handle1:CAS:528:DyaK1cXivVaiur8%3D Occurrence Handle9562984
S Kojima T Hashimoto M Hasegawa S Murata S Ohta H Seki N Okada (1993) ArticleTitleClose phylogenetic relationship between Vestimentifera (tube worms) and Annelida revealed by the amino acid sequence of elongation factor-1α. J Mol Evol 37 66–70 Occurrence Handle1:CAS:528:DyaK3sXlsFKlsLw%3D Occurrence Handle8360920
M Kruse SP Leys IM Muller WEG Muller (1998) ArticleTitlePhylogenetic position of the Hexactinellida within the phylum Porifera based on the amino acid sequence of the protein kinase C from Rhabdocalyptus dawsoni. J Mol Evol 46 721–728 Occurrence Handle1:CAS:528:DyaK1cXjslOntbg%3D Occurrence Handle9608055
S Kumar SB Hedges (1998) ArticleTitleA molecular timescale for vertebrate evolution. Nature 392 917–920 Occurrence Handle1:CAS:528:DyaK1cXjtV2jur8%3D Occurrence Handle9582070
K Kusche T Burmester (2001) ArticleTitleDiplopod hemocyanin sequence and the phylogenetic position of the Myriapoda. Mol Biol Evol 18 1566–1573 Occurrence Handle1:CAS:528:DC%2BD3MXlslOisL4%3D Occurrence Handle11470848
GV Lauder KF Liem (1983) ArticleTitleThe evolution and interrelationships of the actinopterygian fishes. Bull Mus Comp Zool 150 95–197
TH Le D Blair T Agatsuma P-F Humair NJH Campbell M Iwagami TJ Littlewood B Peacock DA Johnston J Bartley D Rollinson EA Herniou DS Zarlenga DP McManus (2000) ArticleTitlePhylogenies inferred from mitochondrial gene orders—A cautionary tale from the parasitic flatworms. Mol Biol Evol 17 1123–1125 Occurrence Handle1:CAS:528:DC%2BD3cXksVOlsrs%3D Occurrence Handle10889225
DT Littlewood PD Olson MJ Telford EA Herniou M Riutort (2001) ArticleTitleElongation factor 1-alpha sequences alone do not assist in resolving the position of the acoela within the metazoa. Mol Biol Evol 18 437–442 Occurrence Handle1:CAS:528:DC%2BD3MXhvVKrt7o%3D Occurrence Handle11230546
DR Maddison WP Maddison (2001) MacClade 4: Analysis of phylogeny and character evolution. Sinauer Associates Sunderland, MA
J Mallatt CJ Winchell (2002) ArticleTitleTesting the new animal phylogeny: First use of combined large-subunit and small-subunit rRNA gene sequences to classify the protostomes. Mol Biol Evol 19 289–301 Occurrence Handle1:CAS:528:DC%2BD38XitFSnsL0%3D Occurrence Handle11861888
AP Martin TM Burg (2002) ArticleTitlePerils of paralogy: Using HSP70 genes for inferring organismal phylogenies. Syst Biol 51 570–587 Occurrence Handle12228000
D McHugh (1997) ArticleTitleMolecular evidence that echiurans and pogonophorans are derived annelids. Proc Natl Acad Sci USA 94 8006–8009 Occurrence Handle1:CAS:528:DyaK2sXksl2ms7s%3D Occurrence Handle9223304
DP Mindell MD Sorenson DE Dimcheff (1998) ArticleTitleMultiple independent origins of mitochondrial gene order in birds. Proc Natl Acad Sci USA 95 10693–10697 Occurrence Handle10.1073/pnas.95.18.10693 Occurrence Handle1:CAS:528:DyaK1cXlvFWhu7o%3D Occurrence Handle9724766
AR Mushegian JR Garey J Martin L-X Liu (1998) ArticleTitleLarge-scale taxonomic profiling of eukaryotic model organisms: A comparison of orthologous proteins encoded by the human, fly, nematode and yeast genomes. Genome Res 8 590–598 Occurrence Handle1:CAS:528:DyaK1cXkt1aru7o%3D Occurrence Handle9647634
C Nielsen N Scharff D Eibye-Jacobsen (1996) ArticleTitleCladistic analyses of the animal kingdom. Biol J Linn Soc Lond 57 385–410
N Nikoh N Iwabe K-i Kuma M Ohno T Sugiyama Y Watanabe K Yasui Z Shi-cui K Hori Y Shimura T Miyata (1997) ArticleTitleAn estimate of divergence time for Parazoa and Eumetazoa and that of Cephalochordata and Vertebrata by aldolase and triose phosphate isomerase clocks. J Mol Evol 45 97–106 Occurrence Handle1:CAS:528:DyaK2sXktl2gu78%3D Occurrence Handle9211740
KJ Peterson DJ Eernisse (2001) ArticleTitleAnimal phylogeny and the ancestry of bilaterians: Inferences from morphology and 18S rDNA gene sequences. Evol Dev 3 170–205 Occurrence Handle1:CAS:528:DC%2BD38XisVWms7Y%3D Occurrence Handle11440251
D Posada KA Crandall (1998) ArticleTitleModeltest: Testing the model of DNA substitution. Bioinformatics 14 817–818 Occurrence Handle10.1093/bioinformatics/14.9.817 Occurrence Handle1:CAS:528:DyaK1MXktlCltw%3D%3D Occurrence Handle9918953
Rambaut A (1995) Se-Al: Sequence Alignment Program. Distributed by the author
TA Rawlings TM Collins R Bieler (2001) ArticleTitleA major mitochondrial gene rearrangement among closely related species. Mol Biol Evol 18 1604–1609 Occurrence Handle1:CAS:528:DC%2BD3MXlslOisLg%3D Occurrence Handle11470854
JC Regier JW Shultz (1997) ArticleTitleMolecular phylogeny of the major arthropod groups indicates polyphyly of crustaceans and a new hypothesis for the origin of hexapods. Mol Biol Evol 14 902–913 Occurrence Handle1:CAS:528:DyaK2sXlvVejurs%3D Occurrence Handle9287423
JC Regier JW Shultz (1998) ArticleTitleMolecular phylogeny of arthropods and the significance of the Cambrian “explosion” for molecular systematics. Am Zool 38 918–928
JC Regier JW Shultz (2001) ArticleTitleElongation factor-2: a useful gene for arthropod phylogenetics. Mol Phylogenet Evol 20 136–148 Occurrence Handle1:CAS:528:DC%2BD3MXksF2mu7s%3D Occurrence Handle11421654
A Rokas PW Holland (2000) ArticleTitleRare genomic changes as a tool for phylogenetics. Trends Ecol Evol 15 454–459 Occurrence Handle11050348
TM Rose ER Schultz JG Henikoff S Pietrokovski CM McCallum S Henikoff (1998) ArticleTitleConsensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res 26 1628–1635 Occurrence Handle1:CAS:528:DyaK1cXis1OjtLY%3D Occurrence Handle9512532
I Ruiz-Trillo M Riutort DT Littlewood EA Herniou J Baguna (1999) ArticleTitleAcoel flatworms: Earliest extant bilaterian metazoans, not members of Platyhelminthes. Science 283 1919–1923 Occurrence Handle1:CAS:528:DyaK1MXitVanurc%3D Occurrence Handle10082465
I Ruiz-Trillo J Paps M Loukota C Ribera U Jondelius J Baguna M Riutort (2002) ArticleTitleA phylogenetic analysis of myosin heavy chain type II sequences corroborates that Acoela and Nemertodermatida are basal bilaterians. Proc Natl Acad Sci USA 99 11246–11251 Occurrence Handle1:CAS:528:DC%2BD38XmslSmtLc%3D Occurrence Handle12177440
AG Saez R Escalante L Sastre (2000) ArticleTitleHigh DNA sequence variability at the alpha 1 Na/K-ATPase locus of Artemia franciscana (brine shrimp): Polymorphism in a gene for salt-resistance in a salt-resistant organism. Mol Biol Evol 17 235–250 Occurrence Handle1:CAS:528:DC%2BD3cXosFygtQ%3D%3D Occurrence Handle10677846
HA Schmidt K Strimmer M Vingron A von Haeseler (2002) ArticleTitleTREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18 502–504 Occurrence Handle1:CAS:528:DC%2BD38XivFKrsL0%3D Occurrence Handle11934758
R Shao NJ Campbell SC Barker (2001a) ArticleTitleNumerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol Biol Evol 18 858–865 Occurrence Handle1:CAS:528:DC%2BD3MXjtFykt7g%3D
R Shao NJ Campbell ER Schmidt SC Barker (2001b) ArticleTitleIncreased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol Biol Evol 18 1828–1832 Occurrence Handle1:CAS:528:DC%2BD3MXmt12hsLo%3D
JW Shultz JC Regier (2000) ArticleTitlePhylogenetic analysis of arthropods using two nuclear protein-encoding genes supports a crustacean + hexapod clade. Proc R Soc Lond B 267 1011–1019 Occurrence Handle1:CAS:528:DC%2BD3cXltFKgt7s%3D
G Steiner M Muller (1996) ArticleTitleWhat can 18S rDNA do for bivalve phylogeny? J Mol Evol 43 58–70 Occurrence Handle1:CAS:528:DyaK28Xks1Grtbg%3D Occurrence Handle8660430
K Strimmer A von Haeseler (1996) ArticleTitleQuartet puzzling: A quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol 13 964–969 Occurrence Handle1:CAS:528:DyaK28XltlSmsLk%3D
DL Swofford (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sinauer Associates Sunderland, MA
M Thollesson JL Norenburg (2003) ArticleTitleRibbon worm relationships—A phylogeny of the phylum Nemertea. Proc R Soc Lond B 270 407–415 Occurrence Handle1:CAS:528:DC%2BD3sXktVWiu70%3D
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24 4876–4882 Occurrence Handle10.1093/nar/25.24.4876
JM Turbeville DM Pfeifer KG Field RA Raff (1991) ArticleTitleThe phylogenetic status of arthropods, as inferred from 18S rRNA sequences. Mol Biol Evol 8 669–686 Occurrence Handle1:CAS:528:DyaK3MXlslKntbo%3D Occurrence Handle1766363
M von Nickisch-Rosenegk WM Brown JL Boore (2001) ArticleTitleComplete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: Gene arrangements indicate that platyhelminths are eutrochozoans. Mol Biol Evol 18 721–730 Occurrence Handle1:CAS:528:DC%2BD3MXjtFyktr0%3D Occurrence Handle11319256
PJ Waddell Y Cao M Hasegawa DP Mindell (1999a) ArticleTitleAssessing the Cretaceous superordinal divergence times within birds and placental mammals by using whole mitochondrial protein sequences and an extended statistical framework. Syst Biol 48 119–137 Occurrence Handle1:STN:280:DC%2BD38zjslSjtw%3D%3D
PJ Waddell N Okada M Hasegawa (1999b) ArticleTitleTowards resolving the interordinal relationships of placental mammals. Syst Biol 48 1–5 Occurrence Handle1:STN:280:DC%2BD38zjslSjsQ%3D%3D
DY-C Wang S Kumar SB Hedges (1999) ArticleTitleDivergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc R Soc Lond B 266 163–171 Occurrence Handle1:CAS:528:DyaK1MXht1Gns7g%3D
WC Wheeler P Cartwright CY Hayashi (1993) ArticleTitleArthropod phylogeny—A combined approach. Cladistics 9 1–39
WC Wheeler M Whiting QD Wheeler JM Carpenter (2001) ArticleTitleThe phylogeny of the extant hexapod orders. Cladistics 17 113–169 Occurrence Handle10.1006/clad.2000.0147
S Whelan N Goldman (2001) ArticleTitleA general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18 691–699 Occurrence Handle1:CAS:528:DC%2BD3MXjtFyktr4%3D Occurrence Handle11319253
K Wilson V Cahill E Ballment J Benzie (2000) ArticleTitleThe complete sequence of the mitochondrial genome of the crustacean Penaeus monodon: Are malacostracan crustaceans more closely related to insects than to branchiopods? Mol Biol Evol 17 863–874 Occurrence Handle1:CAS:528:DC%2BD3cXjvFygs74%3D Occurrence Handle10833192
B Winnepenninckx T Backeljau R De Wachter (1995) ArticleTitlePhylogeny of protostome worms derived from 18S rRNA sequences. Mol Biol Evol 12 641–649 Occurrence Handle1:CAS:528:DyaK2MXmsVOqs7Y%3D Occurrence Handle7659019
B Winnepenninckx T Backeljau R DeWachter (1996) ArticleTitleInvestigation of molluscan phylogeny on the basis of 18S rRNA sequences. Mol Biol Evol 13 1306–1317 Occurrence Handle1:CAS:528:DyaK28XnsVWhtLc%3D Occurrence Handle8952075
J Zrzavy S Mihulka P Kepka A Bezdek D Tietz (1998) ArticleTitlePhylogeny of the Metazoa based on morphological and 18S ribosomal DNA evidence. Cladistics 14 249–285
GR Zug LJ Vitt JP Caldwell (2001) Herpetology: An introductory biology of amphibians and reptiles. Academic Press San Diego
Acknowledgements
The initial phases of this work were performed while F.E.A. and M.T. were postdoctoral fellows in the Division of Invertebrate Zoology working at the Laboratory of Molecular Systematics at the National Museum of Natural History. We would like to thank Jon Norenburg and David Swofford for supporting and assisting this research. We would also like to thank Chris Huddleston and Lynn Atkinson for assistance with various aspects of this work; Allison Gong, Doug Eernisse, Cheryl Dean, and Kerstin Wasson for assistance with specimen collection; Peter Foster and the Natural History Museum (London) for access to computational facilities; the SIUC Office of Research Development and Administration and Department of Zoology for financial assistance; and Natalie Heck and two anonymous reviewers for comments on the manuscript. Species identifications and confirmations were performed by David A. Nickle (Research Entomologist, Systematic Entomology Laboratory, Agriculture Research Service, U.S. Department of Agriculture) for Doru lineare, Natalia J. Vandenberg (Entomologist, USDA Systematic Entomology Laboratory) for Harmonia axyridis, and Joseph Beatty (Emeritus Professor of Zoology, SIUC) for Scolopocryptops sexspinosa. This work was supported in part by NSF Grant DEB-0235794 to F.E.A.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anderson, F.E., Córdoba, A.J. & Thollesson, M. Bilaterian Phylogeny Based on Analyses of a Region of the Sodium–Potassium ATPase β-Subunit Gene . J Mol Evol 58, 252–268 (2004). https://doi.org/10.1007/s00239-003-2548-9
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2548-9