
Abstract
The sixth and eighth steps of histidine biosynthesis are catalyzed by an imidazole glycerol-phosphate (IGP) dehydratase (EC 4.2.1.19) and by a histidinol-phosphate (HOL-P) phosphatase (EC 3.1.3.15), respectively. In the enterobacteria, in Campylobacter jejuni and in Xylella/Xanthomonas the two activities are associated with a single bifunctional polypeptide encoded by hisB. On the other hand, in Archaea, Eucarya, and most Bacteria the two activities are encoded by two separate genes. In this work we report a comparative analysis of the amino acid sequence of all the available HisB proteins, which allowed us to depict a likely evolutionary pathway leading to the present-day bifunctional hisB gene. According to the model that we propose, the bifunctional hisB gene is the result of a fusion event between two independent cistrons joined by domain-shuffling. The fusion event occurred recently in evolution, very likely in the proteobacterial lineage after the separation of the γ- and the β-subdivisions. Data obtained in this work established that a paralogous duplication event of an ancestral DDDD phosphatase encoding gene originated both the HOL-P phosphatase moiety of the E. coli hisB gene and the gmhB gene coding for a DDDD phosphatase, which is involved in the biosynthesis of a precursor of the inner core of the outer membrane lipopolysaccharides (LPS).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Histidine biosynthesis is one of the best-characterized anabolic pathways. There is a large body of genetic and biochemical information, including operon structure, gene expression, and an increasingly larger number of sequences available for this route. This pathway has been extensively studied, mainly in Escherichia coli and Salmonella typhimurium, in both of which the details of histidine biosynthesis are identical (Winkler 1987; Alifano et al. 1996). In all histidine-synthesizing organisms the pathway is unbranched and includes a number of complex and unusual biochemical reactions. It consists of nine intermediates, all of which have been described, and of eight distinct proteins that are encoded by eight genes, hisGDCBHAF(IE), arranged in a compact operon whose complete nucleotide sequence has been determined by Carlomagno et al. (1988). As previously reported (Lazcano et al. 1992; Fani et al. 1995, 1998) there are several independent indications of the antiquity of the histidine biosynthetic pathway suggesting that the entire route was assembled long before the appearance of the Last Universal Common Ancestor (LUCA) of the three extant cell domains. The detailed analysis of the structure and organization of the his genes in (micro)organisms belonging to different phylogenetic archaeal, bacterial, and eucaryal lineages revealed that at least three molecular mechanisms played an important role in shaping the pathway, that is, gene elongation, paralogous gene duplication(s), and gene fusion (Fani et al. 1995; Alifano et al. 1996). The latter has been recognized as one of the major events of gene evolution in the histidine biosynthetic pathway. Such fusions have occurred in the genomes of both Bacteria and some eukaryotes, leading to longer genes encoding bi- or multifunctional enzymes. From this point of view, the hisB gene represents a very interesting case since in S. typhimurium and E. coli it codes for a bifunctional enzyme possessing both histidinol-phosphate phosphatase (EC 3.1.3.15; HOL-Pase) and imidazole glycerol-phosphate dehydratase (EC 4.2.1.19; IGPase) activities responsible for the sixth and the eighth steps of histidine biosynthesis (Winkler 1987). It is widely accepted that the two enzymatic activities of the hisB gene product are associated with two independent domains in the protein, namely, the proximal domain (hisB px ), encoding the phosphatase moiety, and the distal one (hisB d ), encoding the dehydratase (activity) (Fig. 1). This is supported by several independent biochemical and genetic lines of evidence (Loper 1961; Brady and Houston 1973; Chumley and Roth 1981). The structural organization of the two enzymatic activities in some microorganisms supports the two-domain model discussed above. In Saccharomyces cerevisiae the two activities are encoded by two separate genes, HIS2 (for phosphatase activity) and HIS3 (for dehydratase activity) (Broach 1981). The same is true for the his-1 and his-4 genes in Neurospora crassa (Fink 1964). Genes homologous to the S. cerevisiae HIS3 gene have been isolated from other eukaryotes. A similar two-gene organization is also present in some bacterial branches, such as S. coelicolor, in which the two activities are encoded by two different genes (Limauro et al. 1990). The same situation took place in the ancestors of other bacteria and also in archaea, even if the counterpart of the promoter-proximal region (hisB px ) of the E. coli hisB gene has not often been identified (Fani et al. 1995). It has been argued that the HIS2 and HIS3 yeast genes evolved through a “split” mechanism from the prokaryotic domains of the E. coli bifunctional hisB (Glaser and Houston 1974). However, an alternative view has been proposed (Fani et al. 1989, Fani et al. 1995), according to which the evolution of the hisB gene in E. coli and S. typhimurium involved the fusion of two independent cistrons (hisB px and hisB d ), coding for a HOL-Pase and an IGPase, respectively. In spite of the available dataset of genes encoding for HOL-Pase or IGPase, the analyses of these genes carried out until now did not allow us to depict a clear phylogenetic pattern or to trace the evolutionary pathway leading to the extant mono- or bifunctional hisB genes.

Schematic representation of the organization of histidine biosynthetic genes in B. subtilis and E. coli; the HOL-Pase and IGPase activities are encoded by two separate cistrons in B. subtilis and by a bifunctional gene in E. coli. Black circle, IGP-dehydratase; hatched square, HOL-Pase PHP-type; white square, HOL-Pase DDDD-type.
Therefore, the aim of this work was to depict a plausible model for the evolution of hisB genes and to provide insights on the evolution of histidine biosynthetic genes. For this purpose a deep and exhaustive analysis of all the available hisB gene products was carried out.
Materials and Methods
Sequence Retrieval
Amino acid sequences were retrieved from the GenBank, EMBL, and PIR databases. BLAST probing of the protein databases was performed with the BLASTP and Psi-BLAST programs (Altschul et al. 1997) using default parameters. The 16S rDNA sequences were downloaded at the Ribosomal Database Project site (Cole et al. 2003).
Sequence Alignment
The ClustalW program (Thompson et al. 1994) in the BioEdit package (Hall 1999) was used for pairwise and multiple amino acid sequences alignment and shading using default gap penalties and the Dayhoff substitution matrix.
Phylogenetic Tree Construction
Phylogenetic trees were obtained with the MEGA 2.1 software (Kumar et al. 2001), using the neighbor-joining (NJ), minimum evolution (ME), and maximum parsimony (MP) methods. The Poisson correction and the gamma distance models were used for Distance Option in protein sequence analysis as described by Nei and Kumar (2000); the Kimura (1980) two-parameter model was chosen in nucleotide sequence analysis.
Results and Discussion
The Structure of Genes Encoding HOL-Pase and IGPase in the Three Cell Domains
Sequences related to E. coli HisB were retrieved from protein databases using the BLASTP option of the BLAST program (Altschul et al. 1997). This search retrieved at very low E-values (Table 1) 15 sequences showing a high degree of similarity spanning over the entire length of the E. coli protein sequence. As reported in Table 1, all of them belong to proteobacteria, 14 coming from organisms of the γ-subdivision and the remaining 1 is derived from the ε-proteobacterium Campylobacter jejuni. A second, larger group of retrieved sequences exhibited a high degree of similarity only to the phosphatase or the dehydratase moiety of the E. coli HisB protein (not shown). The ClustalW (Thompson et al. 1994) multiple alignment of the bifunctional HisB sequences with the phosphatase and dehydratase moieties from bacteria, where the two enzymatic activities are encoded by separate cistrons, revealed that the domain order was the same in all the bifunctional sequences analyzed, with HOL-Pase located upstream of IGPase, and that the fusion point between the two moieties was identical (not shown). The narrow phylogenetic distribution of HisB bifunctional enzymes (Table 1 and Fig. 2) supported the previous hypothesis (Fani et al. 1989, Fani et al. 1995) that the evolution of the hisB gene in E. coli involved the fusion of two independent cistrons, hisB px and hisB d , coding for a HOL-Pase and an IGPase, respectively. Moreover, the lack of a significant degree of sequence similarity between the two moieties of the bifunctional HisB proteins (not shown) suggested that domain shuffling, rather than gene duplication or gene elongation, was responsible for its present-day structure.

Phylogenetic tree based on 16S rDNA sequences from γ-proteobacteria showing the phylogenetic distribution of the HisB bifunctional enzyme (solid branches). The tree was constructed with the MEGA 2.1 software using the neighbor-joining method, Poisson correction, complete deletion of gaps, and 3025 bootstrap replicates (values above 65% are indicated).
Finally, the fusion event appeared to show a clear phylogenetic pattern, suggesting that it took place recently in an ancestor of some γ-proteobacteria, after the divergence from the β branch. The existence of a bifunctional HisB enzyme in C. jejuni might be the result of a horizontal transfer from a donor γ-proteobacterium possessing a bifunctional enzyme. This hypothesis is supported by the analysis of the (bifunctional) HisB tree (Fig. 3) showing that the C. jejuni sequence falls in the γ-proteobacteria cluster, very close to the Buchnera aphidicola HisB sequence. To check the possibility that other his genes were horizontally transferred from a γ-proteobacterium to C. jejuni, a phylogenetic tree for each of the His proteins was constructed. The phylogenetic trees calculated for the products of hisA and hisD genes are reported in Fig. 3. The topology of these trees is similar to that obtained with HisB, with C. jejuni falling within γ-proteobacteria. This was also true for the trees calculated for the other his gene products (not shown). The analysis of his gene order, structure, and organization in C. jejuni revealed that they are arranged in two compact clusters whose relative gene order, hisGDBHA and hisF(IE), resembles the enterobacterial one. Moreover, the C. jejuni genome harbors a bifunctional hisIE gene, another gene fusion that, within proteobacteria, is peculiar of the enteric lineage and the Xanthomonas/Xylella group. This body of data supports the idea that the C. jejuni hisB bifunctional gene and very likely (at least) most of the other his genes have been horizontally acquired from a γ-proteobacterium. If this lateral inheritance occurred, it could have been facilitated by the fact that all these proteobacteria share similar ecological niches, in most cases represented by the digestive apparatus of some animals. The lack of a HisB bifunctional enzyme in bacteria belonging to the genus Pseudomonas is discussed below.

Phylogenetic tree based on HisA, HisD, and HisB bifunctional amino acid sequences. The P. aeruginosa HOL-Pase and IGPase sequences were fused and used as an outgroup. The tree was constructed with the MEGA 2.1 software using the neighbor-joining method, Poisson correction, complete deletion of gaps, and 5000 bootstrap replicates (values above 65% are indicated).
In the light of data reported in this section showing that the bifunctional hisB genes are the result of a fusion event involving two independent cistrons, we propose a new and unambiguous nomenclature for these genes. Accordingly, hisB px and hisB d will be renamed hisN and hisB, respectively; therefore, the bifunctional hisB gene will be renamed hisNB.
Origin and Evolution of Proteobacterial Bifunctional hisNB Genes
The scenario presented above implies the existence of monofunctional genes encoding HOL-Pase (hisN) and IGP-ase (hisB) in all (micro)organisms with the exclusion of those listed in Table 1. Moreover, if we accept the hypothesis of an ancient origin of the histidine biosynthetic route, it is then plausible to hypothesize the presence of such genes in the genome of the LUCA. These genes might have undergone different evolutionary fates in different cell domains and/or in different phylogenetic lineages within the same domain. In order to depict an evolutionary pathway leading to the extant hisNB genes, the two moieties were analyzed separately.
Evolution of the IGP-Dehydratase Domain
When the archaeal, bacterial, and eucaryal IGP-dehydratase sequences encoded by mono- or bifunctional genes were used as a query in a BLASTP and Psi-BLAST probing of protein databases, they were mutually retrieved at E-values ranging from e −108 to e −4; no other sequence was retrieved at an E-value lower than 0.038. This suggested that the orthologous genes encoding the IGP-dehydratase activity in the extant (micro)organisms are all descendants of an ancestral gene, whose product performed the IGP dehydration and was very likely present in the genome of the LUCA. The lack of sequences related to the IGP-dehydratase reflects the absence in the genome of the extant (micro)organisms of IGP-ase gene paralogs, raising the question of whether this gene may represent a “starter type,” i.e., a gene that was not originated by gene duplication (Lazcano and Miller 1996).
Evolution of the HOL-P Phosphatase Domain
If the evolution of IGP dehydratase encoding genes is simple, the origin and the evolution of HOL-Pase encoding genes appeared to be more complicated, since very little is known about HOL-Pases in organisms containing monofunctional hisB genes. However, the existence of at least two types of enzymes with HOL-Pase activity, belonging to the PHP or to DDDD superfamily, has been demonstrated (Le Coq et al. 1999; Malone et al. 1994). The representatives of the two superfamilies do not share significant sequence similarity, and they use different catalytic residues to perform their enzymatic activity. The PHP superfamily includes the functionally characterized HOL-Pases of Bacillus subtilis (Le Coq et al. 1999) and S. cerevisiae (Malone et al. 1994), the N-terminal domain of the α-subunit of the bacterial DNA polimerase III, a group of stand-alone archaeal and bacterial proteins with unknown function(s), and the C-terminal domain of the family X DNA polymerase of B. subtilis, Aquifex aeolicus, and Methanobacterium thermoautotrophicum (Aravind and Koonin 1998). The E. coli HOL-Pase is part of a large protein superfamily containing L-2-haloacid dehalogenase folds. These proteins catalyze a wide variety of hydrolytic reactions via a covalent substrate–enzyme intermediate. More restrictively E. coli HOL-Pase is a member of a superfamily of phosphohydrolases (Vance and Wilson 2001) which includes several nonspecific phosphatases, named DDDD for the presence of four motifs each containing an aspartate residue essential for the catalytic activity (Thaller et al. 1998).
An exhaustive analysis of protein databases was carried out (i) to check whether the HOL-Pase moieties (HisN) of all bifunctional HisNB enzymes belong to the DDDD superfamily, (ii) to analyze their phylogenetic distribution, (iii) to identify their closest paralogs(s) (if any), and (iv) to depict a possible pathway for their evolution. For this purpose, the bacterial protein databases were probed with either the B. subtilis PHP HOL-Pase (gi 16080014) or the E. coli HOL-Pase sequence (gi 15802501), corresponding to the first 167 residues of the bifunctional enzyme, using the BLASTP and the Psi-BLAST options of BLAST program ( Aitschul et al. 1997). To avoid any misinterpretation as a consequence of partial sequence data, we considered for this kind of analyses only those organisms whose genome has been completely sequenced.
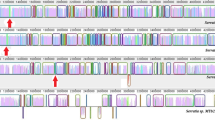
The BLASTP and Psi-BLAST probing with the B. subtilis PHP HOL-Pase did not retrieve any of the proximal domain (HisN) of the 15 available HisNB bifunctional enzymes which were, however, retrieved at E-values ranging from 9e −89to e −38 when the E. coli DDDD HOL-Pase was used as query. This suggested that all 15 HisNB bifunctional enzymes harbored a DDDD HOL-Pase N-terminal moiety and that the appearance of a DDDD-type HOL-Pase was coincident with the appearance of HisNB bifunctional enzymes. This raises the intriguing question of which enzyme performs the HOL-Pase dephosphorylation in bacteria lacking a bifunctional HisNB. To address this issue the sequences retrieved (after three Psi-BLAST iterations) were closely inspected, and this allowed us to find additional bacterial DDDD sequences. Particularly interesting was the discovery that, from the genome of proteobacteria possessing a HisNB protein (with the exception of B. aphidicola, Xanthomonas campestris, X. axonopodis, and Xylella fastidiosa) and from other bacteria (most of which belong to proteobacteria), a DDDD sequence slightly longer than DDDD HOL-Pases (about 190 residues on average) was retrieved at very low E-values (Table 2). In E. coli this DDDD protein is encoded by the gmhB gene (formerly yaeD), which has been functionally characterized (Kneidinger et al. 2002). In this enterobacterium and in other Gram-negative bacteria the GmhB enzyme is involved in the biosynthesis of an ADP-L-β-D-heptose (the activated precursor of a component of the inner core of the outer membrane lipopolysaccharide, LPS) catalyzing the dephosphorylation of D-β-D-heptose 1,7-PP (Kneidinger et al. 2002). Pairwise and multiple ClustalW alignments of HisB phosphatase domain and GmhB proteins (Fig. 4) allowed the detection of a high degree of sequence similarity among (i) the putative GmhB sequences belonging to microorganisms possessing a bifunctional HisB (35–99% identity, 56–99% similarity); (ii) the putative GmhB sequences belonging to microorganisms not possessing a bifunctional HisB (25–34% identity, 46–68% similarity); (iii) the putative GmhB proteins described at points i and ii (26–34% identity, 46–62% similarity); and (iv) the HisN phosphatase domain from the bifunctional enzyme and putative GmhB of the same organism (26–31% identity, 52–57% similarity).

ClustalW alignment of E. coli amino acid sequence HOL-Pase to GmhB sequences from microorganisms possessing a HisB bifunctional enzyme. Gaps were introduced for maximum alignment.
An Evolutionary Model for the Origin and Evolution of Bifunctional hisNB Genes in Proteobacteria
The whole body of data reported in the previous section strongly suggested that HisN, belonging to bifunctional HisNB enzymes, and the putative GmhB protein are encoded by paralogous genes, in that they are the descendants of a common ancestral gene arisen via a duplication event and subsequent evolutionary divergence (Fig. 5). If this is the case, it is possible that the ancestor gene encoded a DDDD-phosphatase with a broad substrate range and able to catalyze (at least) the dephosphorylation of HOL-P and D-β-D-heptose 1,7-PP. Following the duplication event, the two copies underwent an evolutionary divergence that might have narrowed their substrate specificity in such a way that one of them became a HOL-Pase and was then recruited in the histidine biosynthesis, whereas the other copy evolved toward a GmhB protein. In B. aphidicola the absence of gmhB as well as all the other genes for the biosynthesis of cell surface components, including lipopolysaccharides and phospholipids, might reflect the symbiotic lifestyle of this bacterium (Shigenobu et al. 2000). A similar loss might have occurred in both Xylella and Xanthomonas; accordingly, these bacteria (and the others where a gmhB gene is absent) also lack the two other genes (rfaD and rfaE) that in Gram-negative bacteria are involved in the biosynthesis of an ADP-L-β-D-heptose (Kneidinger et al. 2002).

Evolutionary model reconstructing the pathway leading to hisB bifunctional genes in γ-proteobacteria.
The duplication event of the gene encoding the ancestral DDDD protein very likely took place in the γ branch of proteobacteria after its separation from the β branch. This would agree with the finding that β-proteobacteria possess a DDDD phosphatase encoded by a gene similar to gmhB, but they lack a “DDDD”-type and a PHP-type HOL-Pase (Table 2). If this scenario is correct, with the HOL-Pase encoding gene evolved through a duplication event of an ancestral DDDD gene, one might expect that in those bacteria lacking a bifunctional HisB enzyme, but possessing a GmhB-like protein, this might have retained its original functions, that is (at least), the HOL-Pase and GmhB activities. Therefore, one could argue that in a phylogenetic tree containing HOL-Pase, GmhB and the putative GmhB-like proteins (aspecific GmhB) should form three independent clusters. Data concerning this issue are reported in Fig. 6 and are in agreement with the proposed model. A further support to this idea relies on data reported in Table 3, showing the degree of sequence similarity between the E. coli RfaE, GmhB, and RfaD and their orthologs from P. aeruginosa (which possesses a putative aspecific GmhB but not a characterized HOL-Pase), Y. pestis, and H. influenzae. As expected for a model involving a putative aspecific GmhB protein, the P. aeruginosa GmhB showed a degree of similarity to the E. coli ortholog much lower than that found when RfaE and RfaD were compared; additionally, the Y. pestis and H. influenzae GmhB proteins showed a similarity value to the E. coli ortholog similar to that exhibited by the respective RfaE and RfaD proteins.

Phylogenetic tree of GmhB, putative “aspecific” GmhB, and HOL-Pase domains of HisB bifunctional enzymes. The tree was constructed with the MEGA 2.1 software using the neighbor-joining method, Poisson correction, complete deletion of gaps, and 4025 bootstrap replicates (values are indicated above branches).
The existence of an ancestral gene encoding for an aspecific DDDD-type phosphatase is in agreement with the so-called “patchwork” hypothesis (Ycas 1974; Jensen 1976) proposed to explain the origin and evolution of metabolic pathways. In this hypothesis, metabolic pathways have been assembled through the recruitment of primitive enzymes that could react with a wide range of chemically related substrates. Such relatively slow, nonspecific enzymes may have enabled primitive cells containing small genomes to overcome their limited coding capabilities. According to this idea, an ancestral enzyme endowed with low substrate specificity might be able to bind different substrates and catalyze different, though similar reactions. Single or multiple paralogous duplication(s) of the gene encoding the aspecific enzyme and the subsequent divergence of the new sequence(s) led to the appearance of enzymes showing a diversification of functions and a narrowing of specificity. In this way, an enzyme belonging to a given metabolic route might be “recruited” to serve a novel pathway. The patchwork theory is supported by the broad substrate (Jensen 1976) specificity of several contemporary enzymes, which can catalyze classes of chemical reactions, by sequence comparison of paralogous genes, and by the “directed-evolution” experiments in which a microbial (typically prokaryotic) population is subjected to a selective pressure, leading to the establishment of new phenotypes capable of exploiting different substrates (Clarke 1974; Mortlock and Gallo, 1992; Peretò et al. 1998, Jurgens et al. 2000).
The existence of the hisN–hisB gene fusion in the genome of γ-proteobacteria is not an isolated example; additional gene fusions occurred in these genomes, such as that involving the hisI and hisE genes. It is noteworthy that most of the bifunctional proteins recognized to date are involved in metabolic pathways of the γ-subdivision of proteobacteria (Jensen and Ahmad 1990). Even though there is no reason to think that these organisms are more prone to gene fusions than any others, it is interesting that these gene fusions appeared to be parallel to the increasing compactness of the his and other operons. Actually, the analysis of the organization of his genes in bacteria revealed that all 15 hisNB genes are embedded within compact operons, whereas monofunctional genes encoding HOL-Pase are in most cases located outside the histidine gene clusters. This is not so surprising if we agree on the existence of aspecific phosphatases that might perform the HOL-P dephosphorylation. Indeed, it is plausible that the expression of a gene whose product catalyzes more than one chemical reaction in different metabolic pathways should be constitutively expressed rather than being controlled by mechanisms specific for a single route. This is supported by at least two lines of evidence: (i) the mold N. crassa possesses a constitutive alkaline phosphatase that can efficiently use L-histidinol-phosphate as substrate (Morales et al. 2000); (ii) in B. subtilis the HOL-P ase activity is carried out by a PHP-type phosphatase encoded by a his gene whose transcription is not repressed by histidine or histidinol (Le Coq et al. 1999) (Fig. 1). Interestingly, B. subtilis possesses another gene involved in histidine biosynthesis coding for an enzyme able to perform similar reactions in different metabolic pathways and that is located, as the gene coding the HOL-Pase activity, outside the his operon (Fig. 1). This gene, hisC, encodes not only the histidine-pathway transaminase but also an aromatic-pathway transaminase function.
The different organization and localization of hisN, hisB, and bifunctional hisNB genes raise the question of the timing of the fusion event in relation to the building-up of the compact his proteobacterial operons. The availability and analysis of fully sequenced genomes have revealed that operon organization is not a general feature in the microbial world and that an operon instability can be detected even in close phylogenetic lineages. This is also true for the histidine biosynthetic genes. Bacteria belonging to different subdivisions (or to different genera of the same subdivision) within the proteobacterial branch often exhibit different his gene organizations, with most γ-proteobacteria showing overall the most compact organization (Brilli et al. 2002). Organisms belonging to the α- or β-subdivisions show a different organization, with some genes clustered together and others scattered throughout the genome (Fani et al. 1995; Brilli et al. 2002). In spite of this, a certain “order” can be recognized in histidine gene clusters, since in most bacteria where at least some his genes are grouped, four of them are often clustered and arranged in the same relative order (hisBHAF) (see Fig. 5), even in phylogenetically distant bacteria (Fani et al. 1995; Alifano et al. 1996). This cluster corresponds to the so-called “core” of the histidine biosynthesis (Fani et al. 1995), since the encoded proteins catalyze the three sequential steps responsible for the interconnection of histidine biosynthesis to the nitrogen metabolism and the de novo synthesis of purynes. This suggests that the compact his operons might have been assembled by adding either a single his gene or a combination of them to this “core.” It is noticeable that in the γ-proteobacteria P. aeruginosa and P. putida the his genes are localized into three different his clusters, in which they are arranged in the same relative order (hisGDC, hisBHAF, hisIE) (see also Fig. 5) as in the enterobacterial complete his operon, hisGDC(NB) HAF(IE) (Carlomagno et al. 1988).
If this hypothesis is correct, at least two possible scenarios can be depicted for the joining of hisN to his gene clusters/operons, after its evolutionary divergence from the ancestral aspecific DDDD phosphatase encoding gene (Fig. 5). The first one predicts that the DDDD gene copy evolving toward a HOL-Pase might have been inserted in an already formed compact his operon to place the HOL-Pase encoding gene under the same transcriptional control of the other his biosynthetic genes and to coordinate the biosynthesis of all the His enzymes (Fig. 5, Scenario 1). The alternative view (Fig. 5, Scenario 2) would imply that the recruitment of one of the two DDDD paralogs was parallel to the building-up of the entire and compactors his operon. In other words, if we assume that the enterobacterial his operon originated by a progressive addition of his genes to the hisBHAF cluster, one could imagine that the hisN gene joined the hisBHAF “core” by positioning itself just upstream of hisB. Then the (eventual) intervening sequence between hisN and hisB would have been lost and further DNA rearrangements would have led to the fusion of the two moieties. Subsequently, the other his genes completed the operon. We favor the first scenario on the basis of two lines of evidence: (i) the existence in E. coli and S. typhimurium of a functional transcription promoter of the σ70 class, referred to as p2, located just upstream of the bifunctional hisNB gene (Alifano et al. 1996), which might represent the vestige of the original promoter controlling the expression of the ancestral gene; and (ii) most importantly, the finding that, while in some bacteria some his genes are clustered to form the “core” of histidine biosynthesis (hisBHAF), similar clusters including also the hisN moiety have not been recognized so far.
The lack of a hisNB gene in microorganisms belonging to the genus Pseudomonas is parallel to the lack of his operon compactness (see above). It is possible that the organization of the his genes in Pseudomonas might reflect that of the γ-proteobacterial ancestor and that, for still unclear reasons, the assembling of the his operon did not occur in organisms belonging to this genus. On the other hand, an alternative and equally possible explanation is that bacteria belonging to the genus Pseudomonas lack the fusion because the lineage had diverged prior to the recent origin of the γ-proteobacterial fusion. If this is so, we should imagine that the Xanthomonas/Xylella hisNB fusion originated by lateral gene transfer (LGT). This second scenario is strongly supported by at least three lines of evidence: (i) the Xanthomonas/Xylella, His proteins are close to the enteric orthologs in a phylogenetic tree (Fig. 3); (ii) the his gene order and organization are identical to the enterobacterial one (not shown); and (iii) bacteria belonging to these genera possess a bifunctional hisIE gene, which is peculiar of enterobacteria and their relatives (not shown). Therefore, the hisN–hisB fusion might be traced in the ancestor of some proteobacteria rid after their separation from Pseudomonas.
Finally, the fusion of the two domains (HOL-P phosphatase and IGP dehydratase) might have been evolutionary selected and fixed to ensure a fixed ratio of gene products which belong to the same biochemical pathway, i.e., to obtain the coordinate synthesis of the two enzymatic activities. Apparently natural gene fusions that link unrelated pathways are not known. Spatial “channeling” of intermediates is another frequently proposed benefit (Jensen and Ahmad 1990; Fani et al. 1998).
The LUCA Harbored Different Phosphatases to Catalyze HOL-P Dephosphorylation
The existence of (at least) two different phosphatases performing the HOL-Pase activity raises the question of whether the LUCA harbored monofunctional genes coding for DDDD or PHP phosphatase or both of them. For this reason, the phylogenetic distribution of DDDD- and PHP-type phosphatases with a putative HOL-Pase activity was traced by probing the protein databases with (i) the E. coli HOL-Pase domain, (ii) the B. subtilis HOL-Pase (gi 16080014), and (iii) the S. cerevisiae HOL-Pase sequences (gi 14318548), using the BLASTP and Psi-BLAST options of the BLAST program (Altschul et al. 1997).
Data obtained can be summarized as follows.
(i) In addition to the bacterial sequences described in the previous sections, the E. coli HOL-Pase sequence retrieved archaeal, eucaryal, and other bacterial sequences, suggesting a wide distribution of DDDD phosphatase encoding genes. Some of the archaeal retrieved sequences exhibited a significant degree of sequence similarity to the bacterial HOL-Pases (average identity value, 26%), suggesting that (at least) in some Archaea the enzyme responsible for the HOL-P dephosphorylation might belong to the DDDD superfamily. This idea is also supported by the apparent absence of PHP-type HOL-Pases in archaeal organisms (see below). However, the possibility that the HOL-P dephosphorylation might be achieved by other types of phosphatase cannot be ruled out. Furthermore, none of the 49 eucaryal sequences retrieved after three Psi-BLAST iterations exhibited a significant degree of sequence similarity to the E. coli HOL-Pase, suggesting that these phosphatasic domains are very likely involved in other metabolic pathways rather than in histidine biosynthesis. These findings are also supported by the existence of the S. cerevisiae HIS2 protein which does not show any sequence similarity to DDDD HOL-Pase and has been functionally characterized as a PHP phosphatase.
(ii) The B. subtitles PHP HOL-Pase permitted us to retrieve an inventory of 100 sequences, after three Psi-BLAST iterations. No archaeal sequence was retrieved at significant E-values, suggesting that it is not involved in histidine biosynthesis (see above), although the PHP domain is present in some Archaea both as stand-alone proteins and as a domain within polymerases (Aravind and Koonin 1998). Among the bacterial retrieved sequences, most of them belonged to low-GC Gram-positive bacteria. Concerning the eucaryal domains, only the S. cerevisiae and S. pombe sequences were retrieved at significant E-values. Accordingly, the BLAST probing of protein databases retrieved only PHP-type HOL-Pases. If the entire his biosynthetic pathway is ancient and was assembled before the appearance of LUCA, then the latter should have harbored all the genes involved in histidine biosynthesis, including those coding for enzymes with a HOL-Pase activity. On the other hand, data obtained showed that the evolutionary pathway of HOL-Pases leading to the extant mono- and bifunctional hisB genes is complex. The universal phylogenetic distribution of the two types of phosphatases (DDDD and PHP) and their lack of sequence similarity could reflect the existence of two different ancestral nonspecific phosphatases (Le Coq et al. 1999) in the genome of the LUCA, in addition to genes encoding phosphatases other than DDDD- or PHP-type. According to Jensen’s (1976) hypothesis on the origin and evolution of metabolic pathways, these phosphatases would have been slow and inefficient but able to perform the dephosphorylation of a wide range of substrates. It is possible that one (or more) of them would have also been able to perform the HOL-P dephosphorylation and that for a long time this activity would have been carried out by aspecific phosphatases. The substrate specialization of one of these phosphatases toward a narrowed HOL-Pase appeared to be a relatively recent event, at least in bacteria where a complex scenario may be depicted. Data reported in Table 2 revealed that both functionally characterized and putative PHP-type phosphatases were confined to low-GC Gram-positive bacteria. An analogous restricted phylogenetic distribution, limited to proteobacterial harboring HisNB bifunctional enzymes, was observed also for specific DDDD-phosphatases responsible for HOL-P dephosphorylation (Table 1 and 2). Very likely, other bacteria still use less specific DDDD- or other phosphatases to perform HOL-P dephosphorylation.
To our knowledge, no biochemical, genetic, or functional data concerning the archaeal HOL-Pase is available; nevertheless, the analyses carried out in this work indicated that in these microorganisms, a DDDD-type, and very likely other phosphatases, might have been recruited in histidine biosynthesis, whereas the PHP-type phosphatase domain was recruited in other metabolic pathways. A different situation occurred in Eucarya, in which no DDDD-type phosphatases exhibiting a high degree of sequence similarity to either E. coli HOL-Pase or GmhB was found. In some eucaryotes, phosphatases belonging to the PHP superfamily and other aspecific phosphatases are apparently responsible for the dephosphorylation of HOL-P.
Conclusion
In conclusion, we believe that our data have shed some light on the mechanisms responsible for the building up of the histidine biosynthetic pathway. In general, the availability of an increasingly larger body of information regarding gene structure and organization allows us to refine evolutionary models and to shed light on the molecular mechanisms responsible for the assembly of metabolic pathways. This is underscored by the fact that the previous hypothesis predicting the possibility that the enterobacterial bifunctional hisNB (formerly hisB) gene might have been the outcome of a fusion event involving two separate cistrons (hisN and hisB) occurred recently in evolution (Fani et al. 1989) has been confirmed by the analysis of a large number of completely sequenced cellular genomes. Moreover, data reported in this work allowed us to refine and clarify the evolutionary history of the hisN (formerly hisB px ), hisB (formerly hisB d ), and hisNB genes. According to the model proposed in this work, the fusion event leading to the extant hisNB genes may be more precisely traced within γ-proteobacteria after the separation of Pseudomonas. Then the bifunctional hisNB gene, very likely together with all the other his genes, has been laterally transferred to other proteobacteria, such as C. jejuni, Xanthomonas, and Xylella. Our data also revealed that hisN and gmhB are paralogous genes, originated via a duplication event of a gene encoding a phosphatase with a broad range of substrate specificity and located outside the his operons/clusters. It is interesting that all the hisN genes identified so far belong to very compact operons. If Scenario 1 reported in Fig. 5 (predicting that after the duplication from the ancestral DDDD gene, hisN positioned itself within an already constructed histidine operon) is correct, then the lack of hisN outside of compact operons suggests that its introgression within the ancestral his operon (upstream of hisB) might have been positively selected since it permitted completion of the operon and placement of all the genes required for histidine biosynthesis under the same transcriptional regulatory control mechanism(s). It is also possible that the hisN introgression and its successive fusion to hisB might have occurred in a relative short evolutionary time in a sort of “gene duplication–gene fusion coupling.”
The evolutionary history of hisN, hisB, and hisNB, and the paralogy between hisN and gmhB give additional important support to Jensen’s (1976) hypothesis on the origin and evolution of metabolic pathways, strengthening the idea that gene duplication, gene fusion, and recruitment of genes encoding enzymes with a broad range of substrate specificity played a crucial role in the assembly of the entire histidine biosynthetic pathway (Fani et al. 1995, 1998)).
References
P Alifano R Fani P Liò A Lazcano M Bazzicalupo MS Carlomagno CB Bruni (1996) ArticleTitleHistidine biosynthetic pathway and genes: Structure, regulation and evolution. Microbiol Rev 60 44–69 Occurrence Handle1:CAS:528:DyaK28XitVyrs7c%3D Occurrence Handle8852895
SF Altschul TL Madden AA Schäffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25 3389–3402 Occurrence Handle9254694
L Aravind EV Koonin (1998) ArticleTitlePhosphoesterase domains associated with DNA polymerase of diverse origins. Nucleic Acids Res 26 IssueID16 3746–3752 Occurrence Handle10.1093/nar/26.16.3746 Occurrence Handle1:CAS:528:DyaK1cXls1SmtLk%3D Occurrence Handle9685491
DR Brady LL Houston (1973) ArticleTitleSome properties of the catalytic sites of imidazoleglycerolephosphate dehydratase-histidinol posphate phosphatase, a bifunctional enzyme from Salmonella typhimurium. J Biol Chem 248 2588–2592 Occurrence Handle1:CAS:528:DyaE3sXhsFKjs7c%3D Occurrence Handle4349042
M Brilli A Lazcano P Liò R Fani (2002) ArticleTitleStructure and evolution of the histidine biosynthetic pathway. Orig Life Evol Biosph 32 488
JR Broach (1981) Genes of Saccharomyces cerevisiae. JN Strathern EW Jones JR Broach (Eds) The molecular biology of the yeast Saccharomyces: Life cycle and inheritance. Cold Spring Harbor NY 653–727
MS Carlomagno L Chiarotti P Alifano AG Nappo CB Bruni (1988) ArticleTitleStructure of the Salmonella typhimurium and Escherichia coli K-12 histidine operons. J Mol Biol 203 585–606 Occurrence Handle1:CAS:528:DyaK3cXhtFaqtrc%3D Occurrence Handle3062174
FG Chumley JR Roth (1981) ArticleTitleGenetic fusions that place the lactose genes under histidine operon control. J Mol Biol 145 IssueID4 697–712 Occurrence Handle1:CAS:528:DyaL3MXhvVyntLk%3D Occurrence Handle6267294
PH Clarke (1974) The evolution of enzymes for the utilization of novel substrates. MJ Carlile JJ Skehel (Eds) Evolution in the microbial world. Cambridge University Press Cambridge
JR Cole B Chai TL Marsh RJ Farris Q Wang SA Kulam S Chandra DM McGarrell TM Schmidt GM Garrity JM Tiedje (2003) ArticleTitleThe Ribosomal Database Project (RDP-II): Previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31 IssueID1 442–443 Occurrence Handle10.1093/nar/gkg039 Occurrence Handle1:CAS:528:DC%2BD3sXhvFSls7o%3D Occurrence Handle12520046
R Fani M Bazzicalupo G Damiani A Bianchi C Schipani V Sgaramella M Polsinelli (1989) ArticleTitleCloning of the histidine genes of Azospirillum brasilense: Organization of the ABFH gene cluster and nucleotide sequence of the hisB gene. Mol Gen Genet 216 224–229 Occurrence Handle1:CAS:528:DyaK3cXntFSlsw%3D%3D Occurrence Handle2664449
R Fani P Liò A Lazcano (1995) ArticleTitleMolecular evolution of the histidine biosynthetic pathway. J Mol Evol 41 760–774 Occurrence Handle1:CAS:528:DyaK28XivFOjsQ%3D%3D Occurrence Handle8587121
R Fani E Mori E Tamburini A Lazcano (1998) ArticleTitleEvolution of the structure and chromosomal distribution of histidine biosynthetic genes. OLEB 28 555–570 Occurrence Handle10.1023/A:1006531526299 Occurrence Handle1:CAS:528:DyaK1cXmtVWnsbs%3D
GR Fink (1964) ArticleTitleGene-enzyme relations in histidine biosynthesis in yeast. Science 146 525–527 Occurrence Handle1:CAS:528:DyaF2MXhtVOmsA%3D%3D Occurrence Handle14190241
RD Glaser LL Houston (1974) ArticleTitleSubunit structure and photooxidation of yeast imidazoleglycerolphosphate dehydratase. Biochemistry 13 IssueID25 5145–5152 Occurrence Handle1:CAS:528:DyaE2MXltlaguw%3D%3D Occurrence Handle4611478
TA Hall (1999) ArticleTitleBioEdit: A user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41 95–98 Occurrence Handle1:CAS:528:DC%2BD3cXhtVyjs7Y%3D
RA Jensen (1976) ArticleTitleEnzyme recruitment in evolution of new function. Annu Rev Microbiol 30 409–425 Occurrence Handle10.1146/annurev.mi.30.100176.002205 Occurrence Handle1:CAS:528:DyaE28XlvVyitLs%3D Occurrence Handle791073
RA Jensen S Ahmad (1990) ArticleTitleNested gene fusions as markers of phylogenetic branchpoints in prokaryotes. Trends Ecol Evol 5 IssueID7 219–224 Occurrence Handle10.1016/0169-5347(90)90135-Z
C Jürgens A Strom D Wegener S Hettwer M Wilmanns R Sterner (2000) ArticleTitleDirected evolution of a (βα)8-barrel enzyme to catalyze related reactions in two different metabolic pathways. Proc Natl Acad Sci USA 97 9925–9930 Occurrence Handle10.1073/pnas.160255397 Occurrence Handle10944186
M Kimura (1980) ArticleTitleA simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16 111–120 Occurrence Handle7463489
B Kneidinger C Marolda M Graninger A Zamyatina F McArthur P Kosma MA Valvano P Messner (2002) ArticleTitleBiosynthesis pathway of ADP-L-glycero-β-D-manno-heptose in Escherichia coli. J Bacteriol 184 IssueID2 363–369 Occurrence Handle10.1128/JB.184.2.363-369.2002 Occurrence Handle1:CAS:528:DC%2BD38XjtVyksw%3D%3D Occurrence Handle11751812
S Kumar K Tamura IB Jacobsen M Nei (2001) ArticleTitleMEGA2: Molecular evolutionary genetics analysis software. Bioinformatics 17 IssueID12 1244–1245 Occurrence Handle1:CAS:528:DC%2BD38XmtVCktQ%3D%3D Occurrence Handle11751241
A Lazcano SL Miller (1996) ArticleTitleThe origin and early evolution of life: prebiotic chemistry, the pre-RNA world, and time. Cell 85 793–798 Occurrence Handle1:CAS:528:DyaK28XjslSkurs%3D Occurrence Handle8681375
A Lazcano GE Fox J Oro’ (1992) Life before DNA: The origin and evolution of early Archean cells. RP Mortlock (Eds) The evolution of metabolic function. CRC Press Boca Raton FL 237–339
D Le Coq S Fillinger S Aymerich (1999) ArticleTitleHistidinol phosphate phosphatase, catalyzing the penultimate step of the histidine biosynthesis pathway, is encoded by ytvP (hisJ) in Bacillus subtilis. J Bacteriol 181 IssueID10 3277–3280 Occurrence Handle1:CAS:528:DyaK1MXjt1yrsbc%3D Occurrence Handle10322033
D Limauro A Avitabile C Cappellano AM Puglia CB Bruni (1990) ArticleTitleCloning and characterization of the histidine biosynthetic gene cluster of Streptomyces coelicolor A3(2) Gene 90 IssueID1 31–41 Occurrence Handle10.1016/0378-1119(90)90436-U Occurrence Handle1:CAS:528:DyaK3MXhvVWksw%3D%3D Occurrence Handle2199329
JC Loper (1961) ArticleTitleEnzyme complementation in mixed extracts of mutants from the Salmonella histidine B locus. Proc Natl Acad Sci USA 47 1440–l450 Occurrence Handle1:CAS:528:DyaF38Xislarug%3D%3D Occurrence Handle13763426
RE Malone S Kim SA Bullard S Lundquist L Hutchings-Crow S Cramton L Lutfiyya J Lee (1994) ArticleTitleAnalysis of a recombination hotspot for gene conversion occurring at the HIS2 gene of Saccharomyces cerevisiae. Genetics 137 IssueID1 5–18 Occurrence Handle1:CAS:528:DyaK2cXmvFCjt7g%3D Occurrence Handle8056323
AC Morales SR Nozawa Jr G Thedei Jr W Maccheroni A Rossi (2000) ArticleTitleProperties of a constitutive alkaline phosphatase from strain 74A of the mold Neurospora crassa. Braz J Med Biol Res 33 905–912 Occurrence Handle1:CAS:528:DC%2BD3cXmsFOlsLw%3D Occurrence Handle10920432
RP Mortlock MA Gallo (1992) Experiments in the evolution of gatabolic pathways using modern bacteria. RP Mortlock (Eds) The evolution of metabolic functions. CRC Press Boca Raton, FL 1–13
M Nei S Kumar (2000) Molecular evolution and phylogenetics. Oxford University Press New York
J Peretò R Fani JI Leguina A Lazcano (1998) Enzyme evolution and the development of metabolic pathways. A Cornish-Bowden (Eds) New beer in an old bottle: Eduard Buchner and the growth of biochemical knowledge. Universitat de Valencia Valencia 173–198
S Shigenobu H Watanabe M Hattori Y Sakaki H Ishikawa (2000) ArticleTitleGenome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407 IssueID6800 81–86 Occurrence Handle10.1038/35024074 Occurrence Handle1:CAS:528:DC%2BD3cXmslelu7o%3D
MC Thaller S Schippa GM Rossolini (1998) ArticleTitleConserved sequence motifs among bacterial, eukaryotic, and archaeal phosphatases that define a new phosphohydrolase superfamily. Protein Sci 7 IssueID7 1647–1652 Occurrence Handle1:CAS:528:DyaK1cXks1eqtbk%3D Occurrence Handle9684901
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight-matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
JR Vance TE Wilson (2001) ArticleTitleUncoupling of 3’-phosphatase and 5’-kinase functions in budding yeast. J Biol Chem 276 IssueID18 15073–15081 Occurrence Handle10.1074/jbc.M011075200 Occurrence Handle1:CAS:528:DC%2BD3MXjs1OrsLY%3D Occurrence Handle11278831
ME Winkler (1987) Biosynthesis of histidine. FC Neidhardt JL Ingraham KB Low B Magasanik M Schaechter HD Humbarger (Eds) Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. ASM Press Washington, DC 395–411
M Ycas (1974) ArticleTitleOn the earlier states of the biochemical system. J Theor Biol 44 145–160 Occurrence Handle1:CAS:528:DyaE2cXhtFOhur4%3D Occurrence Handle4207200
Acknowledgements
We are very grateful to three anonymous reviewers for their helpful comments and suggestions on improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brilli, M., Fani, R. Molecular Evolution of hisB Genes . J Mol Evol 58, 225–237 (2004). https://doi.org/10.1007/s00239-003-2547-x
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2547-x