
Abstract
The statistical analysis of phylogenetic footprints in the two known horn shark Hox clusters and the four mammalian clusters shows that the shark HoxN cluster is HoxD-like. This finding implies that the most recent common ancestor of jawed vertebrates had at least four Hox clusters, including those which are orthologous to the four mammalian Hox clusters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hox genes code for homeodomain containing transcription factors which are homologous to the genes in the Drosophila homeotic gene clusters (McGinnis and Krumlauf 1992). Vertebrates, in contrast to all invertebrates examined, have multiple Hox gene clusters that presumably have arisen from a single ancestral cluster in the most recent common ancestor of chordates, i.e., amphioxus and vertebrates (Garcia-Fernández and Holland 1994; Kappen et al. 1989). The timing of the Hox cluster duplication events in vertebrate phylogeny is still somewhat unclear. The most popular hypothesis is that the common ancestor of sharks and bony fish (which also include the land vertebrates such as human and mouse) had four clusters homologous to the mammalian ones. (Holland and Garcia-Fernández 1996). To test this idea, two nearly complete Hox clusters have recently been isolated and sequenced, called N and M (Kim et al. 2000). While the M cluster is clearly homologous to the human HoxA cluster, it was more difficult to assign the homology to the HoxN cluster. In the original description HfHoxN was identified as homologous to the human HoxD cluster, but there is also evidence consistent with homology to the HoxC cluster (Málaga-Trillo and Meyer 2001).
Materials and Methods
In this contribution we perform a statistical analysis of conserved noncoding sequences utilizing a new software called Tracker (Prohaska et al. 2003). This program is based on BLAST (Altschul et al. 1990) for the initial search of all pairs of input sequences. Comparisons are (optionally) restricted to homologous intergenic regions. The resulting list of pairwise sequence alignments is then assembled into groups of partially overlapping regions that are subsequently passed through several filtering steps. Individual phylogenetic footprints (PFs) are defined in Tagle’s original paper (Tagle et al. 1988) as blocks of at least 6 bp of DNA sequence that is 100% conserved in taxa that have an additive evolutionary time of 250 million years. PFs are considered to be putative transcription factor binding sites. Typically Tracker detects clusters of such footprints which are termed cliques. The decomposition of cliques into individual footprints is often ambiguous. Our statistical analysis below is therefore based on the total length of significantly homologous noncoding sequence fragments between pairs of clusters. This measure is roughly proportional to the number of individual footprints. Homologous footprints are necessarily colinear (disregarding the possibility of local transpositions or inversions which cannot be resolved with the present analysis method due to the highly diverged sequence outside the footprint clusters). Noncolinear Tracker hits are therefore disregarded (marked × in the supplemental material).
The Tracker program produces alignments of the footprint cliques using Dialign2 (Morgenstern 1999). These are padded with “gap” characters in those sequences that do not take part in a particular clique and then concatenated. The resulting “alignment” is sparse in the sense that the “gap” character is the most frequent letter. The reconstruction of phylogenies from such a dataset has to take three complications into account. (1) Gene loss will almost certainly cause the loss of all the associated regulatory sequences. In the extreme case, presence-absence data of footprints might just reflect the presence–absence pattern of the genes. (2) We cannot expect to have detected all footprints in all species. (3) Gain and loss of footprints are not symmetric processes: In fact footprint loss is much easier than the de novo creation. These complications can be circumvented by considering only mutations within conserved noncoding regions, i.e., within the footprint cliques detected by the Tracker program. The distance of two clusters is therefore derived from the frequency of mutations within cliques that are shared by the two clusters. Technically, this amounts to treating “gaps” as missing data rather than as an additional character state.
We use different distance-based and parsimony-type approaches here: the neighbor joining method (Saitou and Nei 1987) (implemented in the PHYLIP package, version 3.6) (Felsenstein 1989), canonical split decomposition (Bandelt and Dress 1992), Buneman (1971) trees, parsimony splits, and P-trees (Bandelt and Dress 1993). With the exception of NJ, these methods are implemented in the Splitstree package (version 3.1) (Huson 1998). The split-based methods are particularly suitable for our purposes because they are known to be very conservative in that they tend to produce multifurcations rather than poorly supported edges (Semple and Steel 2003). In addition, we use MacClade 4.0 (Maddison and Maddison 2000) for standard maximum parsimony analysis.
The following sequences are used for the analysis: shark (Heterodontus francisci)—M-cluster HfM = AF479755, N-cluster HfN = AF224263; human (Homo sapies)—HsA = AC004080.2rc + AC010990 [201-6508]rc + AC004079 [75001-end]rc, HsB = NT-010783 [931646-1263780]rc, HsC = NT-009563 [580371-708054]rc, HsD = NT-037537 [4075338-end]; and rat (Rattus norvegicus)—RnA = NW-043751 [910030-1194462]rc, RnB = NW-042671 [264022-581839], RnC = NW-044048 [722873-1060956], RnD = NW-042732 [1061702-1217610]rc. Fugu (Takifugu rubripes) sequences are taken from the Fugu database DOE Joint Genome Institute: TrAa = scaffold_47 of release_3.0, TrAb = scaffold 1874 of release 2.0, and TrD = scaffold_3959+ scaffold_214 [160440-end]rc. Here “rc” means that the reverse complement of the database entry has been used (after extracting the indicated interval).
Results
A comparison of the protein sequences of the shark HoxN cluster with mammalian Hox protein sequences is consistent with D-likeness, although the data in Table 1 do not show an unambiguous picture. In particular, the HoxD proteins are not always the ones with the highest degree of sequence identity; see Table 1. In a similar vein, the analysis of Hox genes and of genes linked to the Hox clusters such as collagens does not yield an unambiguous picture for the branching order of the four mammalian Hox clusters (Bailey et al. 1997).
Let us now turn to the analysis of the conserved parts of the noncoding sequences. Table 2 summarizes the results of pairwise comparisons of shark and human (or rat) Hox clusters. It should be noted that the sequence of the shark HoxN cluster is incomplete, spanning only the sequence from evx to (almost) Hox-4. There is a particularly high conservation of noncoding sequences between shark HoxM and mammalian HoxA sequences in the range from Hox-4 to Hox-1. As a consequence, the counts for HoxN are significantly lower. In Table 2 we therefore display the data for both the full-length clusters and the restriction to the region between evx and Hox-4. The total length of sequences conserved between shark and mammalian clusters in this region is comparable between HfM and HfN.
The homology of the shark HoxM and the mammalian HoxA clusters is obvious from these data. For the HoxN sequence we find only a moderate signal in the numbers of colinear cliques. The total length of the conserved regions, however, is more than twice as large with HoxD as with HoxC and about 50% longer in HoxD compared to HoxA. The location and distribution of the footprint cliques (Fig. 1) also strongly argue for a homology with HoxD rather than HoxC.

Overview of the phylogenetic footprint cliques produced by Tracker for the comparison of the horn shark HoxN sequence (HfN) and the human HoxC (HsC) and HoxD(HsD) sequences, respectively. X denotes the Evx genes.
A comparison of HfHoxN with the fugu (Takifugu rubripes) HoxCα and HoxD sequences also places HfHoxN with the D rather than the C cluster. These data must be interpreted with caution. (i) The Fugu sequences are preliminary constructs combining two or three scaffolds and hence not complete. (ii) Even though the current version 3.0 of the Fugu genome database (DOE Joint Genome Institute) does not contain evidence of a Cβ cluster, it is most likely that the teleost C cluster was duplicated since the zebrafish (Danio rerio) does have both a HoxCα and a HoxCβ cluster (Amores et al. 1998). The duplication event might have caused the additional loss of a substantial number of footprints. Nevertheless, we find that the counts for the shark–pufferfish comparisons are similar to the shark–mammal comparisons.
The sensitivity of the Tracker method is increased by including more sequences. In particular, homologous footprints can be identified between two sequences even if they do not yield a significant signal when the two sequences are compared directly. We have therefore performed a complete analysis of both shark clusters and all four human Hox clusters. The supplemental material lists all footprint cliques in the range from evx to hox-1 that appear in at least one shark and at least one human cluster. The statistics of the conserved regions between clusters is summarized in Table 3.
Treating phylogenetic footprint cliques as pre-sence/absence characters in a parsimony framework also supports the hypothesis that HfHoxN is more closely related to HsHoxD than to HsHoxC. The tree ((A,M),(C,(D,N)) is seven steps shorter than ((A,M),((C,N),D) (tree length = 402, CI = 0.57, RI = 0.18). This result is based on the assumption of a ((A,B),(C,D)) scenario, which is favored from the analysis of Hox sequences; see, for instance, Amores et al. (1998). The alternative, which is supported by the analysis of genes linked to the Hox clusters by Bailey et al. (1997), leads to considerably shorter trees. The tree (((A,M),(D,N)),(C,B)) has 374 steps (CI = 0.62, RI = 0.43) compared to (((A,M),D),((C,N),B)), which is 44 steps longer. While we do not want to get into the question of which cluster phylogeny applies to the human Hox clusters here, we just want to note that in either scenario a tree with HfHoxN most closely related to HsHoxD is more parsimonious than any other phylogenetic position of HfHoxN (data not shown).
These data clearly indicate that the shark HoxN cluster is HoxD-like at least as far as the noncoding sequences are concerned. In fact, based on the total size of the footprints that are shared between clusters, the next candidate would be the mammalian A cluster, not the C cluster as proposed in Málaga-Trillo and Meyer (2001).
To test whether HfHoxN is a true homologue of the mammalian HoxD clusters we consider the cooccurrences of the 49 footprint cliques that are present in HfHoxN (Table 4). In particular, there are 14 cliques that HfHoxN shares uniquely with human HsHoxD, compared to 10 cliques shared with the HoxC cluster and only 5 or 6 that are only shared with the HoxA and HoxB clusters, respectively. On the other hand, about 30% of the footprints are shared between HfHoxN, one of the human clusters, and at least one other mammalian cluster. The footprints shared between HoxN and either HoxA or HoxB are, to 90%, also shared with HoxM. Together, these data strongly suggest that HfHoxN not only is most similar to the mammalian HoxD clusters but is a true homologue.
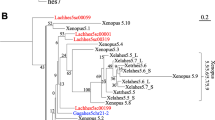
Further evidence for this claim can be obtained from the phylogenetic analysis of the combined footprint cliques of the four mammalian clusters for either human or rat together with the two available shark sequences. Both distance-based (Fig. 2) and parsimony-based methods (Fig. 3) agree on this interpretation. We have chosen a variety of split-based algorithms for this analysis because these techniques are known to produce multifurcations rather than poorly supported edges. For comparison standard neighbor-joining trees are shown in Fig. 2.

Distance-based phytogenies of shark and mammalian Hox clusters. Neighbor joining trees (Saitou and Nei 1987) are computed using Felsenstein’s PHYLIP package (version 3.6). Buneman graphs representing the canonical decomposition of the distance function and the split-based Buneman trees are computed using Daniel Huson’s (1998) Splitstree package, version 3.1.

Parsimony-based phylogenies of shark and mammalian Hox clusters computed using Splitstree, version 3.1 (Huson 1998).
All data presented in Figs. 2 and 3 either support the conclusion that the shark HoxN cluster is homologous with mammalian HoxD cluster or are at least consistent with this conclusion (whenever the HfHoxN–HoxD node is a multifurcation).
Discussion
The evidence presented in this paper supports the original hypothesis, namely, that the shark HoxN cluster is orthologous to the mammalian HoxD cluster (Kim et al.2000). The method employed is novel, namely, to use the distribution and extent of noncoding sequences for phylogenetic inferences. Below we discuss the implications of the present finding for our understanding of Hox cluster evolution in vertebrates.
Conserved noncoding sequences have long been used to find candidate cis-regulatory elements; see Duret and Bucher (1997) for a review. Identification of putative cis-regulatory sequences requires long stretches of sequence from distantly related species (Tagle et al. 1988) or a set of species which have sufficient additive divergence among them (Sumiyama et al. 2001). More recently this method has been used to trace the noncoding sequence divergence after HoxA cluster duplication in teleosts (Chiu et al. 2002). In this paper it has been shown that noncoding sequences can remain highly conserved in the absence of Hox gene cluster duplication, as documented between the shark HoxM and the mammalian HoxA cluster (see also this paper). Hence it is possible to treat the loss and the acquisition of conserved noncoding sequences as potentially apomorphic characters. Thus they contain phylogenetic information. The congruence between the structural and coding sequence evidence and the comparison of noncoding sequence conservation for HoxM and HoxA cluster validates this assumption. In the case of the shark HoxN cluster the evidence from coding sequence and structural organization is less strong and we thus rely on the evidence from noncoding sequence conservation. While the signal is still not as strong as for the HoxM, each analysis is at least consistent and in many cases positively supportive of orthology between the shark HoxN and the mammalian HoxD cluster.
The conclusion that both the shark HoxM and HoxN clusters are directly orthologous to the mammalian HoxA and HoxD clusters, respectively, has important implications for the history of Hox cluster duplications. It follows that the most recent common ancestor of cartilaginous fish and the bony fish clade (which includes mammals) had at least four Hox clusters orthologous to the four mammalian Hox clusters. It is thus likely that sharks have two more clusters than those currently described. This evidence also confirms the hypothesis of Peter Holland that the four-cluster situation typical for most major gnathostome lineages has arisen before the most recent common ancestor of all recent gnathostomes (Garcia-Fernández and Holland 1994; Holland et al. 1994). Of course this result does not guarantee that all gnathostome lineages in fact have at least four Hox clusters since clusters can be lost. This can happen, in particular, soon after the duplication, which might have occurred shortly before the split between the shark and mammalian lineages.
References
SF Altschul W Gish W Miller EW Myers DJ Lipman (1990) ArticleTitleBasic local alignment search tool. J Mol Biol 215 403–410 Occurrence Handle10.1006/jmbi.1990.9999 Occurrence Handle1:CAS:528:DyaK3MXitVGmsA%3D%3D Occurrence Handle2231712
A Amores A Force YL Yan et al. (1998) ArticleTitleZebrafish hox clusters and vertebrate genome evolution. Science 282 1711–1714 Occurrence Handle1:CAS:528:DyaK1cXnslGgtrY%3D Occurrence Handle9831563
S Aparicio J Chapman E Stupka et al. (2002) ArticleTitleWhole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297 1301–1310 Occurrence Handle10.1126/science.1072104 Occurrence Handle1:CAS:528:DC%2BD38Xms1ejtr8%3D Occurrence Handle12142439
WJ Bailey J Kim G Wagner FH Ruddle (1997) ArticleTitlePhylogenetic reconstruction of vertebrate Hox cluster duplications. Mol Biol Evol 14 843–853 Occurrence Handle1:CAS:528:DyaK2sXltVeisbw%3D Occurrence Handle9254922
HJ Bandelt AWM Dress (1992) ArticleTitleA canonical decomposition theory for metrics on a finite set. Adv Math 92 47
HJ Bandelt AWM Dress (1993) A relational approach to split decomposition. O Opitz B Lausen R Klar (Eds) Information and classification. Springer-Verlag Berlin 123–131
P Buneman (1971) The recovery of trees from measures of dissimilarity. FR Hodson DG Kendall P Tauto (Eds) Mathematics and the archeological and historical sciences. Edinburgh University Press Edinburgh, UK 387–395
Ch Chiu C Amemiya K Dewar CB Kim FH Ruddle GP Wagner (2002) ArticleTitleMolecular evolution of the HoxA cluster in the three major gnathostome lineages. Proc Natl Acad Sci USA 99 5492–5497 Occurrence Handle10.1073/pnas.052709899 Occurrence Handle1:CAS:528:DC%2BD38XjtFKltrw%3D
DOE Joint Genome Institute (2002) Fugu genome database. version 2.0: http://genome.jgi-psf.org/fugu3/fugu3.home.html ; version 3.0: http://genome.jgi-psf.org/fugu6/fugu6.home.html
L Duret P Bucher (1997) ArticleTitleSearching for regulatory elements in human noncoding sequences. Curr Opin Struct Biol 7 399–406
J Felsenstein (1989) ArticleTitlePHYLIP—Phylogeny inference package (version 3.2). Cladistics 5 164–166
J Garcia-Fernández PW Holland (1994) ArticleTitleArchetypal organization of the amphioxus hox gene cluster. Nature 370 563–566 Occurrence Handle1:CAS:528:DyaK2cXmslensLY%3D Occurrence Handle7914353
PW Holland J Garcia-Fernández (1999) ArticleTitleHox genes and chordate evolution. Dev Biol 173 382–395 Occurrence Handle10.1006/dbio.1996.0034
PWH Holland J Garcia-Fernández NA Williams A Sidow (1994) ArticleTitleGene duplication and the origins of vertebrate development. Development . IssueID(Suppl) 125–133
DH Huson (1998) ArticleTitleSplitstree: Analyzing and visualizing evolutionary data. Bioinformatics 14 68–73
C Kappen K Schughart FJH Ruddle (1989) ArticleTitleTwo steps in the evolution of antennapedia-class vertebrate homeobox genes. Proc Natl Acad Sci USA 86 5459–5463 Occurrence Handle1:CAS:528:DyaL1MXkvFWmu74%3D Occurrence Handle2568634
CB Kim C Amemiya W Bailey K Kawasaki J Mezey W Miller S Minosima N Shimizu GP Wagner GP Ruddle (2000) ArticleTitleHox cluster genomics in the horn shark, Heterodontus francisci. Proc Nat Acad Sci USA 97 1655–1660 Occurrence Handle10.1073/pnas.030539697 Occurrence Handle1:CAS:528:DC%2BD3cXhsFCrsbg%3D Occurrence Handle10677514
DR Maddison WP Maddison (2000) MacClade 4: Analysis of phylogeny and character evolution. Sinauer Associates Sunderland, MA (e-book and computer program)
E Málaga-Trillo A Meyer (2001) ArticleTitleGenome duplications and accelerated evolution of Hox genes and cluster architecture in teleost fishes. Am Zool 41 6767–686
W McGinnis R Krumlauf (1992) ArticleTitleHomeobox genes and axial patterning. Cell 68 283–302 Occurrence Handle1:CAS:528:DyaK38XhtVShs7o%3D Occurrence Handle1346368
B Morgenstern (1999) ArticleTitleDIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15 211–218 Occurrence Handle10.1093/bioinformatics/15.3.211 Occurrence Handle1:CAS:528:DyaK1MXjs1artrc%3D Occurrence Handle10222408
Prohaska S, Fried C, Flamm C, Wagner G, Stadler PF (2003) Surveying phylogenetic footprints, in large gene clusters: Applications to Hox cluster duplications. Mol Phylog Evol (in press)
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
C Semple M Steel (2003) Phylogenetics. Oxford University Press Oxford
K Sumiyama C Kim FH Ruddle (2001) ArticleTitleAn efficient cis-element discovery method using multiple sequence comparisons based on evolutionary relationships. Genomics 71 260–262 Occurrence Handle10.1006/geno.2000.6422 Occurrence Handle1:CAS:528:DC%2BD3MXptleqtw%3D%3D Occurrence Handle11161821
DA Tagle BF Koop M Goodman JL Slightom DL Hess RT Jones (1988) ArticleTitleEmbryonic epsilon and gamma globin genes of a prosimian primate (galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol 203 439–455 Occurrence Handle1:CAS:528:DyaL1MXktlCnsrs%3D Occurrence Handle3199442
JD Thompson DG Higgs TJ Gibson (1994) ArticleTitleCLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties, and weight matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
Acknowledgements
Funding for this research is gratefully acknowledged: DFG Bioinformatics Initiative BIZ-6/1-2 to S.J.P., C.F., and P.F.S., NSF IBN-9905408 to F.R. and IBN-0321470 to G.P.W.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prohaska, S.J., Fried, C., Amemiya, C.T. et al. The Shark HoxN Cluster Is Homologous to the Human HoxD Cluster . J Mol Evol 58, 212–217 (2004). https://doi.org/10.1007/s00239-003-2545-z
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2545-z