
Abstract
Halophila stipulacea is a dioecious marine angiosperm, widely distributed along the western coasts of the Indian Ocean and the Red Sea. This species is thought to be a Lessepsian immigrant that entered the Mediterranean Sea from the Red Sea after the opening of the Suez Canal (1869). Previous studies have revealed both high phenotypic and genetic variability in Halophila stipulacea populations from the western Mediterranean basin. In order to test the hypothesis of a Lessepsian introduction, we compare genetic polymorphism between putative native (Red Sea) and introduced (Mediterranean) populations through rDNA ITS region (ITS1-5.8S-ITS2) sequence analysis. A high degree of intraindividual variability of ITS sequences was found. Most of the intragenomic polymorphism was due to pseudogenic sequences, present in almost all individuals. Features of ITS functional sequences and pseudogenes are described. Possible causes for the lack of homogenization of ITS paralogues within individuals are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Halophila stipulacea (Forssk.) Aschers. is a subtidal dioecious marine angiosperm, originally distributed along the western coasts of the Indian Ocean and in the Red Sea. This species was first recorded in the eastern Mediterranean Sea in 1895 (Fritsch 1895) and only since 1988 in the western part of the basin (Villari 1988). The species was initially considered as a tropical relict in the eastern Mediterranean Sea (Pérès 1967), while the current opinion is that H. stipulacea came from the Red Sea after the opening of the Suez Canal in 1869 (Lessepsian hypothesis [Lipkin 1975]).
The genetic and morphological variability was investigated in two western Mediterranean populations of H. stipulacea using RAPD-PCR markers (Procaccini et al. 1999). High genetic variability within and between the two populations was found.
In the present work, we used a nuclear rDNA region (ITS1–5.8S–ITS2) as molecular marker to assess the genetic relationships between Mediterranean Sea and Red Sea populations of H. stipulacea, in order to verify the hypothesis of a recent introduction in the Mediterranean basin. A Lessepsian scenario would result in a weak or absent phylogeographic signal, due to the recent disjunction of populations and repeated migration events, due to antropic activities (i.e., shipping).
ITS has been widely used in phylogenetic and phylogeographic studies on a variety of organisms (Baldwin et al. 1995), possessing features that allow addressing phylogenetic questions from infraspecific to interspecific level (Hillis and Dixon 1991). The whole sequence is constituted by regions with different evolutionary rates: the conserved 5.8S rRNA gene flanked by the two fast evolving internal transcribed spacers, ITS1 and ITS2. ITS1 and ITS2 possess a stable and compact secondary structure and have a functional role maintaining sequences for proper excision in processing the RNA precursor (van Nues et al. 1995; Reed et al. 2000). The ITS region is present with hundreds of tandemly repeated copies within the genome of a single individual and, as part of a multigene family, is subjected to concerted evolution (Dover 1986). Concerted evolution allows homogenization between multiple copies within the genome of a single individual through DNA recombination mechanisms, such as gene conversion (Arnheim 1983) and unequal crossing-over (Smith 1976). If lack of recombination between copies occurs, intragenomic polymorphism can be revealed. rDNA intraindividual heterogeneity is now recognized to be a common feature and has been described in various animals and plant species (Suh et al. 1993; Buckler and Holtsford 1996a, b; Campbell et al. 1997; Serrão et al. 1999; Denduangboripant and Cronk 2000; Famà et al. 2000; Harris and Crandall 2000; Kita and Ito 2000; Reed et al. 2000; Coyer et al. 2001; Gandolfi et al. 2001; Hartmann et al. 2001; Muir et al. 2001).
An alternative source of intragenomic polymorphism could be the presence of pseudogenic sequences. Pseudogenes are copies of the transcriptional unit that undergo to a loss of function, with subsequent relaxing of selective constraints and accumulation of mutations. The presence of pseudogenes for the ITS region has been recently described in various plant and animal species (Buckler and Holtsford 1996a, b; Hartmann et al. 2000; Muir et al. 2001; Kita and Ito 2000; Hughes et al. 2002; Márquez et al. 2003).
In this paper, genetic polymorphism of the ITS region in H. stipulacea is described from the interpopulation to the intraindividual level. The presence of a subset of ITS sequences presenting pseudogenic characteristics with increased evolution rates with respect to the functional ones is also described.
Materials and Methods
Specimen Collection
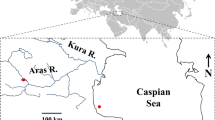
Individuals were collected by SCUBA diving from three locations, two in the Red Sea (Ras Mohammed and Small Creek, Egypt; two individuals each) and one in the Mediterranean basin (one individual from Rhodes Island, Greece). Four individuals from Vulcano Island (Sicily, Italy) sampled for a previous work (Procaccini et al. 1999) were also included in the analysis (Fig. 1).

Sampling sites. Red Sea: 1, Ras Mohammed (Egypt); 2, Small Creek (Egypt); Mediterranean Sea: 3, Rhodes Island (Greece); 4, Vulcano Island (Sicily, Italy).
DNA Extraction and PCR Amplification
DNA was extracted using the CTAB method of Doyle and Doyle (1987) modified as by Procaccini et al. (1996). Amplification of the complete ITS region (ITS1-5.8S-ITS2) was performed in an Omn-E thermal cycle (Hybaid) using the universal primers ITS1 and ITS4 (White et al. 1990). The amplification profile consisted of a denaturation step of 4 min at 94°C followed by 30 cycles as follows: 1 min at 94°C, 1 min at 58°C, and 2 min at 72°C. A final extension step of 7 min at 72°C was added. Amplification products were gel-purified after separation on 1.5% agarose, with the QiaexII Gel Extraction kit (Qiagen).
Cloning and Sequencing
First attempts at direct sequencing were not successful, due to multiple peaks per position in the electropherograms. PCR products were then cloned using the TOPO-TA cloning kit (Invitrogen).
Two to seven clones for each individual (38 in total) were sequenced on a Beckman CEQ 2000 Automated Sequencer using the Dye-Terminator cycle sequencing kit (Beckman).
Sequence Alignment and Analysis
Alignment was conducted using BioEdit 4.5.8 software (Hall 1999) and checked manually. Phylogenetic reconstruction for the entire ITS region (ITS1, 5.8S, and ITS2) was conducted using the MEGA software (version 2.1 [Kumar et al. 2001]). Analyses were performed using the Kimura two-parameter distance method with 1000 bootstrap replicates. A phylogenetic tree was constructed using the neighbor-joining method. Halophila ovalis was included in the analysis and used as an outgroup (sequence obtained by Michelle Waycott, James Cook University, Townsville, Australia).
Analysis of haplotype and nucleotide diversity was conducted using DNAsp software (Rozas and Rozas 1999), according to Nei (1987). P i values were calculated using a sliding window approach (window width: 30 bp).
Hierarchical analysis of variance (AMOVA) was conducted using Arlequin software (Schneider et al. 2000), considering the (i) intraindividual, (ii) among-individual, and (iii) among-population level.
In order to characterize putative pseudogenic sequences, the following analyses were performed. A relative rate test was conducted using the two-cluster test of Takezaki et al. (1995) included in the Phyltest software, version 2.0 (Kumar 1996), where the constancy of the molecular clock is examined for two lineages when an outgroup lineage is given. If L a and L b are the averages of observed numbers of substitutions per site (branch lengths) from the common ancestor of clusters A and B, then L a = L b is the null hypothesis under constancy of the molecular clock, i.e., δ = L a − L b = 0, and δ will become negative if lineage B is evolving faster than A. Deviation of δ from 0 was tested by a two-tailed normal deviate test, with a Z value >1.96 required to reject rate constancy at the 5% level. We tested putative pseudogenic sequences (cluster B) against functional sequences (cluster A), considering only one copy for each haplotype. H. ovalis was used as an outgroup. The Kimura two-parameter model was used as the distance estimation method.
Methylation-related substitution sites (C→T and G→A) were checked by eye, considering as putative methylation sites dinucleotides CdG and trinucleotides CdNdG (Gardiner-Garden et al. 1992). The GC content for each sequence was determined by BioEdit software.
Free energy of secondary structures for the three regions was calculated using mFold (Zucker 1989).
Results
Thirty-eight sequences were obtained by cloning from nine specimens collected in the four populations (see Fig. 2 for population codes).

Neighbor-joining tree constructed on the Kimura two-parameter distance matrix (1000 bootstrap replicates). Populations are in capital letters, numbers identify the individuals, and lowercase letters indicate different clones. Populations: RM, Ras Mohammed; SC, Small Creek; VU, Vulcano Island; RH, Rhodes Island. Arrows indicate pseudogenic sequences.
Twenty-two haplotypes were found of 38 sequences (GenBank accession numbers AY352600–AY352637). Substitutions and indels appeared to be distributed evenly all along the repeat units. Five mononucleotidic indels were present; one 4-bp deletion was present in the ITS2 subregion in one sequence (SC2e) and two large deletions of 31 bp were found in the same subregion in two other sequences (RM1f and RM2e). Sequences presented the following characteristics: ITS1 was 218–221 bp long, presenting 54 polymorphic sites, a nucleotide diversity (P i ) of 0.025, and a haplotype diversity of 0.839; the 5.8S region was 161–163 bp long, having 27 polymorphic sites, P i of 0.012, and a haplotype diversity of 0.570; ITS2 was 194–226 bp long, presenting 29 polymorphic sites, a P i of 0.010, and a haplotype diversity of 0.422. Polymorphism values ranked as follows: ITS1 > 5.8S > ITS2.
A neighbor-joining tree inferred from pairwise Kimura two-parameter distances on the whole alignment showed a lack of phylogeographic pattern, with low bootstrap values. Clones from single individuals, in fact, were scattered across the tree (Fig. 2). The AMOVA conducted for ITS1, 5.8S, and ITS2 separately confirmed that most of the variability was due to intraindividual polymorphism (Table 1a).
A group of eight sequences (marked in the tree with arrows) showed higher levels of polymorphism. These sequences were in a basal position within the phylogenetic tree (see Fig. 2). The average number of nucleotide differences between these eight sequences (henceforth called set 2) and the remainder of the sequences (set 1) was 19.833. The comparison between P i values of the two sets showed that divergence within set 2 is much higher than divergence within set 1 (Fig. 3). Some sequences from set 2 showed large deletions (4-bp deletion in SC2e clone and 31-bp deletion in RM1f and RM2e clones).

Nucleotide diversity (P i ) for set 1 and set 2 sequences. Subdivision among regions is also shown.
Results of an AMOVA on set 1 showed that most of the variability was still at the intraindividual level (Table 1b). Variance was higher for the complete set of sequences than for set 1 only. The 5.8S region showed the lowest variance.
The high level of variation found in the sequences in set 2 was considered a clue for the possible presence of pseudogenes. In order to verify this hypothesis the following analyses were performed.
-
1
A relative rate test based on Kimura two-parameter distance was conducted to assess differential substitution rates among set 1 and set 2 sequences (Table 2). For all regions, rate constancy was rejected at the 5% level. The substitution rate of the set 1 subregions is ITS1 > 5.8S ≅ ITS2. Rates for set 2 sequences maintain the same ranking, with values significantly higher than those obtained for the set 1 sequences.
-
2
As a result of relaxed selective constraints, pseudogenes are often characterized by an elevated rate of deamination-like substitutions, related to methylation sites (Gojobori et al. 1982; Li et al. 1984). Deamination-driven mutations, related to putative methylation sites (dinucleotides CdG and trinucleotides CdNdG [Gardiner-Garden et al. 1992]), were compared between functional and putative pseudogenic sequences. In Table 3 the number of G→A and C→T methylation-related mutations is shown for putative pseudogenes and for functional sequences in comparison with nonmethylation sites. The greatest number of mutations was found in ITS1 regions, the lowest in the 5.8S region. As a result of the high deamination rates, the average GC content in pseudogenes is significantly lower than in the functional sequences. Values are within the range of those reported in the literature for angiosperms (50–75% in ITS1 and ITS2 [Baldwin et al. 1995]).
-
3
In order to investigate if putative pseudogenes presented a lower stability of their secondary structure, as expected from the loss of functional constraints, we measured the free energy (ΔG) of the RNA secondary structure for the three subregions separately (Table 3). Stability of the RNA secondary structure was significantly lower in pseudogenes than in functional sequences for ITS1 and ITS2 (P < 0.05), while differences were not significant for the 5.8S.
In order to verify if the presence of paralogous pseudogenes in the data set had determined the lack of resolution in the phylogeographic pattern obtained, we also constructed phylogenetic trees separately for functional sequences and pseudogenes. Lack of phylogeographic signal was confirmed in both cases (data not shown).
Discussion
The aim of the present work was to verify the hypothesis of a Lessepsian origin for the presence of H. stipulacea in the Mediterranean Sea. The Lessepsian hypothesis implies that the Mediterranean populations of H. stipulacea established after the migration of individuals from the Red Sea through the Suez Canal, thus representing a subsample of the genetic pool of the native populations. We sampled two populations in the original areal (Red Sea) and two populations in the Mediterranean Sea, one from the eastern and one from the western part of the basin. Using a nuclear ribosomal DNA region (ITS1–5.8S–ITS2), no clear differentiation was observed, either between Mediterranean and Red Sea or within the Mediterranean populations, supporting the hypothesis of a recent Lessepsian introduction. The observed absence of any phylogeographic pattern between native (Red Sea) and introduced (Mediterranean) populations of H. stipulacea, in fact, suggests a recent disjunction and a continuous and intensive gene flow through recruitment of new polymorphic individuals from the native areal, possibly as a consequence of human activities (e.g., shipping through the Suez Canal).
The alternative hypothesis of Pérès (1967) that considers H. stipulacea as a tropical relict would have resulted in a sensible genetic divergence between populations from the two studied areals.
Our study also reports the existence of intraindividual polymorphism in the ITS region in H. stipulacea. The rDNA multigene family is known to undergo concerted evolution, resulting in homogenization among unit copies through gene conversion (Arnheim 1983) and unequal crossing-over (Smith 1976) at an estimated rate of 10−2–10−4 events per kilobase per generation (Dover 1989). The degree of homogenization depends on the effectiveness of these mechanisms in the face of novel mutations, which allow spreading of new variants in populations. An increasing number of recent papers reports intraindividual variability for this region, mostly as a consequence of recent hybridization events (Suh et al. 1993) and multiple NORs (nucleolar organizing regions) on nonhomologous chromosomes (Suh et al. 1993; Jellen et al. 1994).
Information about the ploidy level and number and location of NORs in H. stipulacea is unfortunately lacking, but we hypothesize that the ITS intraindividual polymorphism found could be the consequence of two contrasting factors.
On one hand, concerted evolution could have failed because of loss of recombination through sexual reproduction (Campbell et al. 1997). Although at least gene conversion is known to occur both meiotically and mitotically, mitotic processes seem to be much less effective than meiotic ones (Jinks-Robertson and Petes 1993). Rare sexual recombination could be supported, at least in the Mediterranean populations, by the rarity of female flowers, recorded only recently for the first time in the basin (Di Martino et al. 2000).
On the other hand, in the presence of a high level of gene flow, new variants can be introduced and spread within a population at rates faster than their homogenization (Sanderson and Doyle 1992; Hartmann et al. 2001). The latter hypothesis is more likely, considering H. stipulacea as an introduced species characterized by high levels of migration between native and colonized areas.
Most of the intraindividual variability found was due to the presence of putative ITS pseudogenic sequences in the genome of H. stipulacea. ITS pseudogenes have been described in various plant species, such as Zea mays (Buckler and Holtsford 1996a, b), the genus Lophocereus (Hartmann et al. 2001), the genus Quercus (Muir et al. 2001), the genus Aconitum (Kita and Ito 2000), and the genus Leucaena (Hughes et al. 2002), and recently in a marine organism, the coral genus Acropora (Márquez et al. 2003).
Pseudogenic features, with respect to functional units, can be summarized as follows: (i) a higher evolutionary rate; (ii) a higher number of methylation-related mutations due to deamination (C→T and G→A) and therefore a lower GC content; and (iii) a higher free energy of the secondary structure of the transcript, resulting in a lower stability. All these features are consequences of the loss of selective constraints implying that, under the neutral mutation hypothesis, the fate of new mutations is determined almost completely by genetic drift (Li et al. 1984).
Our analyses showed that 8 of the 23 haplotypes found (34.8%) present pseudogenic characteristics. This high representation of pseudogenes in the cloned fragments of the studied genome could be explained through a preferential PCR amplification due to the lower GC content of these sequences, which allows easier denaturation of the DNA fragment during the PCR cycling (Wagner et al. 1994). Eventual contamination or amplification of organellar DNA was checked through a BLAST search (http://www. ncbi.nlm.nih.gov/BLAST/ ) and excluded.
Although differences in stability of secondary structure between 5.8S functional and pseudogenic sequences seem not to be statistically significant, rates of evolution for 5.8S putative pseudogenes were much higher with respect to the functional ones, indicating that processes leading to the formation of pseudogenes have been acting in these sequences. High levels of ITS intragenomic variability has been shown in few marine photosyntetic organisms (e.g., Caulerpa [Famà et al. 2000], Fucus [Serrão et al. 1999], Macrocystis [Coyer et al. 2001]), while the only other marine angiosperm in which low ITS intraindividual polymorphism has been described is Posidonia oceanica (Capiomont, unpublished data).
The ever-growing evidence of the presence of pseudogenic sequences for the ITS region could lead to a reconsideration of the observed intragenomic variability in marine photosynthetic organisms. More studies are needed in order to verify if the presence of pseudogenes in H. stipulacea represents a particular feature of this species or if it is also characteristic of other phylogenetically related seagrass species.
In conclusion, the present study is the first report of the presence of high intraindividual polymorphism and pseudogenes in the rDNA of a marine angiosperm and represents another indication that cautions must be taken when using ITS regions in phylogenetic studies.
References
N Arnheim (1983) Concerted evolution in multigene families. M Nei R Koehn (Eds) Evolution of genes and proteins. Sinauer Sunderland, MA
BG Baldwin MJ Sanderson JM Porter MF Wojciechowski CS Campbell MJ Donoghue (1995) ArticleTitleThe ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Ann Ma Bot Garden 82 247–277
ES IV Buckler TP Holtsford (1996a) ArticleTitle Zea systematics: Ribosomal ITS evidence. Mol Biol Evol 13 612–622 Occurrence Handle1:CAS:528:DyaK28XhvFyisbc%3D
ES IV Buckler TP Holtsford (1996b) ArticleTitle Zea ribosomal repeat evolution and substitution patterns. Mol Biol Evol 13 623–632 Occurrence Handle1:CAS:528:DyaK28XitVOit70%3D
CS Campbell MF Wojciechowski BG Baldwin LA Alice MJ Donoghue (1997) ArticleTitlePersistent nuclear ribosomal DNA sequence polymorphism in the Amelanchier agamic complex (Rosaceae). Mol Biol Evol 14 81–90 Occurrence Handle1:CAS:528:DyaK2sXjvFOqsw%3D%3D Occurrence Handle9000756
JA Coyer GJ Smith RA Andersen (2001) ArticleTitleEvolution of Macrocystis spp. (Phaeophyceae) as determined by ITS1 and ITS2 sequences. J Phycol 37 574–585 Occurrence Handle10.1046/j.1529-8817.2001.037001574.x
J Denduangboripant QCB Cronk (2000) ArticleTitleHigh intra-individual variation in internal transcribed spacer sequences in Aeschynanthus (Gesneriaceae): Implications for phylogenetics. Proc R Soc Lond B 267 1407–1415 Occurrence Handle10.1098/rspb.2000.1157 Occurrence Handle1:CAS:528:DC%2BD3cXmsVSit78%3D Occurrence Handle10983824
V Di Martino G Marino MC Blundo (2000) ArticleTitleQualitative minimal area of a community with Halophila stipulacea from south-eastern Sicilian Coasts (Ionian Sea). Biol Mar Medit 7 677–679
GA Dover (1986) ArticleTitleMolecular drive in multigene families: How biological novelties arise, spread and are assimilated. Trends Genet 2 159–165 Occurrence Handle10.1016/0168-9525(86)90211-8 Occurrence Handle1:CAS:528:DyaL2sXltFWhsr8%3D
GA Dover (1989) ArticleTitleLinkage disequilibrium and molecular drive in the rDNA gene family. Genetics 122 249–252 Occurrence Handle1:CAS:528:DyaL1MXktlChsbo%3D Occurrence Handle2731732
JJ Doyle JL Doyle (1987) ArticleTitleA rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19 11–15
P Famà JL Olsen WT Stam G Procaccini (2000) ArticleTitleHigh levels of intra- and inter-individual polymorphism in Caulerpa racemosa (Forssk.). J. Agardh. Eur J Phycol 35 349–356 Occurrence Handle10.1017/S0967026200002948
C Fritsch (1895) ArticleTitleÜber die auffindung einer marinen Hydrocharidee im Mittelmeer. Verh Zool Bot Ges Wien 45 104–106
A Gandolfi P Bonilauri V Rossi P Menozzi (2001) ArticleTitleIntraindividual and intraspecies variability of ITS1 sequences in the ancient asexual Darwinula stevensoni (Crustacea: Ostracoda). Heredity 87 449–455 Occurrence Handle10.1046/j.1365-2540.2001.00927.x Occurrence Handle1:CAS:528:DC%2BD38XivVyitQ%3D%3D Occurrence Handle11737293
M Gardiner-Garden JA Sved M Frommer (1992) ArticleTitleMethylation sites in angiosperm genes. J Mol Evol 34 219–230 Occurrence Handle1:CAS:528:DyaK38XhvVyqsLg%3D
T Gojobori Li Wen-Hsiung D Graur (1982) ArticleTitlePatterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol 18 360–369 Occurrence Handle1:CAS:528:DyaL38Xlt1Wgtrs%3D Occurrence Handle7120431
TA Hall (1999) ArticleTitleBioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 4 95–98
DJ Harris KA Crandall (2000) ArticleTitleIntra-genomic variation within ITS1 and ITS2 of freshwater crayfishes (Decapoda: Cambaridae): Implications for phylogenetic and Microsatellite studies. Mol Biol Evol 17 284–291 Occurrence Handle1:CAS:528:DC%2BD3cXosFynsA%3D%3D Occurrence Handle10677851
S Hartmann JD Nason D Bhattacharya (2001) ArticleTitleExtensive ribosomal DNA genie variation in the columnar cactus Lophocereus. J Mol Evol 53 124–134 Occurrence Handle1:CAS:528:DC%2BD3MXmt1Sjsbk%3D Occurrence Handle11479683
DM Hillis MT Dixon (1991) ArticleTitleRibosomal DNA: Molecular evolution and phylogenetic inference. Q Rev Biol 66 411–453 Occurrence Handle1:STN:280:By2C2M%2FmsFc%3D Occurrence Handle1784710
CE Hughes CD Bailey SA Harris (2002) ArticleTitleDivergent and reticulate species relationships in Leucaena (Fabaceae) inferred from multiple data sources: Insights into polyploid origins and nr DNA polymorphism. Am J Bot 89 1057–1073 Occurrence Handle1:CAS:528:DC%2BD3sXislejtA%3D%3D
EN Jellen RL Phillips HW Hines (1994) ArticleTitleChromosomal localization and polymorphism of ribosomal DNA in oat (Avena spp. Genome 37 23–32 Occurrence Handle1:CAS:528:DyaK2MXhtVGitQ%3D%3D
S Jinks-Robertson TD Petes (1993) ArticleTitleExperimental determination of rates of concerted evolution. Methods Enzymol 224 631–646 Occurrence Handle1:CAS:528:DyaK2cXis1agtbs%3D Occurrence Handle8264416
Y Kita M Ito (2000) ArticleTitleNuclear ribosomal ITS sequences and phylogeny in East Asian Aconitum subgenus Aconitum (Ranunculaceae), with special reference to extensive polymorphism in individual plants. Plant Syst Evol 225 1–13 Occurrence Handle1:CAS:528:DC%2BD3MXhsFGjsrk%3D
S Kumar (1996) PHYLTEST; Phylogenetic hypothesis testing, version 2.0. Pennsylvania State University University Park
S Kumar K Tamura IB Jakobsen M Nei (2001) MEGA2: Molecular Evolutionary Genetics Analysis software, Arizona State University Tempe
W-H Li C-I Wu C-C Luo (1984) ArticleTitleNonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol 21 58–71 Occurrence Handle1:CAS:528:DyaL2MXntlGhuw%3D%3D Occurrence Handle6442359
Y Lipkin (1975) ArticleTitle Halophila Stipulacea, A review of a successful immigration. AQ.BOT 1 203–215 Occurrence Handle10.1016/0304-3770(75)90023-6
LM Márquez DJ Miller JB Mackenzie MJM Van Oppen (2003) ArticleTitlePseudogenes contribute to extreme diversity of nuclear ribosomal DNA in the hard coral Acropora. Mol Biol Evol 20 1077–1086 Occurrence Handle10.1093/molbev/msg122 Occurrence Handle12777522
G Muir CC Fleming C Schlötterer (2001) ArticleTitleThree divergent rDNA clusters predate the species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol Biol Evol 18 112–119 Occurrence Handle1:CAS:528:DC%2BD3MXotVOmsg%3D%3D Occurrence Handle11158370
M Nei (1987) Molecular evolutionary genetics. Columbia University Press New York
JM Pérès (1967) ArticleTitleThe Mediterranean benthos. Oceanogr Mar Biol Annu Rev 5 449–553
G Procaccini RS Alberte L Mazzella (1996) ArticleTitleGenetic structure of the seagrass Posidonia oceanica in the Western Mediterranean: Ecological implications. Mar Ecol Prog Series 140 153–160
G Procaccini S Acunto P Famà F Maltagliati (1999) ArticleTitleMorphological and genetic variability of Mediterranean populations of Halophila stipulacea (Forssk.) Aschers. (Hydrocharitaceae) at different spatial scale. Mar Biol 135 181–189 Occurrence Handle10.1007/s002270050615
KM Reed JD Hackett RB Phillips (2000) ArticleTitleComparative analysis of intra-individual and inter-species DNA sequence variation in salmonid ribosomal DNA cistrons. Gene 249 115–125 Occurrence Handle10.1016/S0378-1119(00)00156-6 Occurrence Handle1:CAS:528:DC%2BD3cXjsVeltLo%3D Occurrence Handle10831845
J Rozas R Rozas (1999) ArticleTitleDnasp version 3: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15 174–175 Occurrence Handle10.1093/bioinformatics/15.2.174 Occurrence Handle1:CAS:528:DyaK1MXisVOksrY%3D Occurrence Handle10089204
MJ Sanderson JJ Doyle (1992) ArticleTitleReconstruction of organismal and gene phylogenies from data on multigene families: Concerted evolution, homoplasy and confidence. Syst Biol 41 4–17
S Schneider D Roessli L Excoffier (2000) Arlequin ver. 2.000: A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva Geneva, Switzerland
EA Serrão LA Alice SH Brawley (1999) ArticleTitleEvolution of the Fucaceae (Phaeophyceae) inferred from nrDNA-ITS. J Phycol 35 382–394 Occurrence Handle10.1046/j.1529-8817.1999.3520382.x
GP Smith (1976) ArticleTitleEvolution of repeated DNA sequences by unequal crossover. Science 191 528–535 Occurrence Handle1:CAS:528:DyaE28Xhs1Srurg%3D Occurrence Handle1251186
YB Suh LB Thien HE Reeve EA Zimmer (1993) ArticleTitleMolecular evolution and phylogenetic implications of internal transcribed spacer sequences of ribosomal DNA in Winteraceae. Am J Bot 80 1042–1055 Occurrence Handle1:CAS:528:DyaK2cXivFCgsbk%3D
N Takezaki A Rzhetsky M Nei (1995) ArticleTitlePhylogenetic test of the molecular clock and linearized trees. Mol Biol Evol 12 823–833 Occurrence Handle1:CAS:528:DyaK2MXns1yqsbg%3D Occurrence Handle7476128
RW van Nues J Venema JMJ Rientjes A Dirks-Mulder HA Raué (1995) ArticleTitleProcessing of eukaryotic pre-rRNA: The role of the transcribed spacers. Biochem Cell Biol 73 789–801 Occurrence Handle1:CAS:528:DyaK28Xit1Klu7c%3D Occurrence Handle8721995
R Villari (1988) ArticleTitleSegnalazioni floristiche italiane: 565. Halophila stipulacea (Forssk.) Aschers. (Hydrocharitaceae). Genere e specie nuovi per l’Italia. Inform Bot Ital 20 672
A Wagner N Blackstone P Cartwright M Dick B Misof P Snow GP Wagner J Barrels M Murtha J Pendleton (1994) ArticleTitleSurveys of gene families using polymerase chain reaction: PCR selection and PCR drift. Syst Biol 43 250–261
TJ White T Bruns S Lee J Taylor (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. MA Innis DH Gelfand JJ Sninsky TJ White (Eds) PCR protocols: A guide to methods and applications. Academic Press New York 315–322
M Zucker (1989) ArticleTitleOn finding all suboptimal foldings of an RNA molecule. Science 244 48–52 Occurrence Handle1:CAS:528:DyaL1MXkt1SnsbY%3D Occurrence Handle2468181
Acknowledgements
Sampling in the Red Sea was conducted by Ferruccio Maltagliati. We thank Wiebe Kooistra for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Valeria Ruggiero, M., Procaccini, G. The rDNA ITS Region in the Lessepsian Marine Angiosperm Halophila stipulacea (Forssk.) Aschers. (Hydrocharitaceae): Intragenomic Variability and Putative Pseudogenic Sequences . J Mol Evol 58, 115–121 (2004). https://doi.org/10.1007/s00239-003-2536-0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2536-0