
Abstract
Chalcone synthase (CHS) is a key enzyme in the biosynthesis of flavonoides, which are important for the pigmentation of flowers and act as attractants to pollinators. Genes encoding CHS constitute a multigene family in which the copy number varies among plant species and functional divergence appears to have occurred repeatedly. In morning glories (Ipomoea), five functional CHS genes (A–E) have been described. Phylogenetic analysis of the Ipomoea CHS gene family revealed that CHS A, B, and C experienced accelerated rates of amino acid substitution relative to CHS D and E. To examine whether the CHS genes of the morning glories underwent adaptive evolution, maximum-likelihood models of codon substitution were used to analyze the functional sequences in the Ipomoea CHS gene family. These models used the nonsynonymous/synonymous rate ratio (ω = d N /d S ) as an indicator of selective pressure and allowed the ratio to vary among lineages or sites. Likelihood ratio test suggested significant variation in selection pressure among amino acid sites, with a small proportion of them detected to be under positive selection along the branches ancestral to CHS A, B, and C. Positive Darwinian selection appears to have promoted the divergence of subfamily ABC and subfamily DE and is at least partially responsible for a rate increase following gene duplication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants of the morning glory genus (Ipomoea) are distributed worldwide and are characterized by a rich diversity of flower colors. For example, the common morning glory (I. purpurea) has white, pink, and blue or dark-blue flowers (Clegg and Durbin 2000). The diversity in flower color is almost certainly due to differences in either the structural or the regulatory genes of the flavonoid biosynthetic pathway (Durbin et al. 1995), which culminates in the production of anthocyanins, the main pigments responsible for flower color. The presence or absence of these pigments affects the coloration of the floral display, which attracts pollinators. The anthocyanin pigments are therefore important to reproductive success (Clegg and Durbin 2000).
The first step in the flavonoid biosynthetic pathway, the formation of naringenin chalcone from malonyl-CoA and p-coumaroyl-CoA, is catalyzed by the enzyme chalcone synthase (CHS). CHS is a typical homodimeric plant polyketide synthase with two subunits of about 43 kDa (Ferrer et al. 1999). The chalcone synthase from Medicago sativa was first crystallized for X-ray diffraction analysis (Ferrer et al. 1999). The three-dimensional structure of alfalfa CHS2 revealed that four chemically reactive residues (Cys164, Phe215, His303, and Asn336), which are conserved in all the known CHS-related enzymes, define the active site. Topologically, three interconnected cavities intersect with these four residues and form the active site of CHS. These cavities include a CoA-binding tunnel, a coumaroyl-binding pocket, and a cyclization pocket. The volume and shape of these cavities govern starter molecule selectivity, polyketide chain length, and the folding and cyclization pathway. Therefore, alternations in the surface topology may affect the substrate specificity and the mode of cyclization reaction.
There is growing evidence that chalcone synthase is closely related to other plant-specific polyketide synthases, including stilbene synthase (STS) (Schöppner and Kindl 1984; Schröder 1997), acridone synthase (ACS) (Lukacin et al. 1999), bibenzyl synthase (BBS) (Preisig-Müller et al. 1995), 2-pyrone synthase (2PS) (Eckermann et al. 1998), and phlorisovalerophenone synthase (PVPS) (Paniego et al. 1999). It is also observed that very few amino acid changes in those proteins may result in shifts in enzyme function. For instance, the 2-pyrone synthase from Gerbera hybrida shares >70% identity with the CHS enzyme from the same plant but forms a triketide from an acetyl-CoA starter and two malonyl-CoA extender units (Eckermann et al. 1998). The functional shift from CHS to 2PS is due to three mutations in residues lining the active site, which substantially reduce the volume of the pockets for substrate binding and cyclization (Ferrer et al. 1999). Stilbene synthase has been proposed to evolve from CHS independently several times over the course of evolution (Tropf et al. 1994). Tropf et al. (1994) used site-directed mutagenesis to demonstrate that only three amino acid changes in a CHS/STS hybrid construct were necessary to obtain STS enzymatic activity, suggesting that in nature very few amino acid changes are required to change the enzymatic function of a CHS gene.
In morning glories, five functional CHS genes (A–E) have been described (Durbin et al. 2000). Based on genetic distances between sequences, the morning glory CHS genes can be divided into two subfamilies. One subfamily, designated ABC and characterized by Durbin et al. (1995), is composed of CHS A, B, and C genes. The second subfamily, designated DE and characterized by Fukada-Tanaka et al. (1997), is composed of CHS D and E genes. Phylogenetic analysis of the Ipomoea CHS gene family revealed that CHS A, B, and C have long branches within the phylogeny, suggesting accelerated rates of nucleotide substitution. In contrast, CHS D and E have short branches (Durbin et al. 2000). At nonsynonymous sites, the CHS ABC subfamily evolved 2.7 times faster than the CHS DE subfamily (Durbin et al. 2000). The catalytic properties of Ipomoea CHS genes have not yet been extensively characterized. However, biochemical analysis of Ipomoea CHS genes revealed that only CHS D and E are capable of catalyzing the condensation reaction that results in naringenin chalcone, while the CHS A and B genes appear to encode enzymes that produce bisnoryangonin but not naringenin chalcone (Clegg and Durbin 2000).
Gene duplication with subsequent diversification among different copies is considered an important mechanism for functional divergence (Ohno 1970; Ohta 1993). The diversification of different copies may be caused by relaxation of functional constraints in redundant genes (Kimura 1983) but may also be driven by positive selection after gene duplication (Ohta 1993). A stringent criterion of positive Darwinian selection in protein evolution is a significantly higher nonsynonymous (d N; amino acid replacing) than synonymous (d S; silent) substitution rate (Li 1997). The rate ratio ω = d N/d S measures the magnitude and direction of selective pressure on a protein, with ω = 1, <1, and >1 indicating neutral evolution, purifying selection, and positive selection, respectively. This criterion has been used to identify a number of cases of adaptive molecular evolution (Yang and Bielawski 2000).
In this study, we investigate the role of both purifying and positive selection in the evolution of the Ipomoea CHS gene family and examine whether the CHS genes of the morning glories underwent adaptive evolution after gene duplication. Maximum-likelihood models of codon substitution were used to analyze the functional sequences in the Ipomoea CHS gene family. These models allow the ω ratio to vary among lineages or sites and can be used to detect changes in selective pressure or operation of positive selection along branches in the phylogeny.
Materials and Methods
Sequence Data
Eighteen Ipomoea CHS genes were obtained from GenBank. The accession numbers are as follows: CHSA (U15946), CHSB (U15947), CHSC (U15949), CHSD (AB001826), and CHSE (AB001827) for I. purpurea; CHSA (U15943), CHSB (U15944), CHSD (AB001818), and CHSE (AB001819) for I. nil; CHSD1 (AB023791) and CHSLF1 (AB037680) for I. batatas; CHSA (U15945) for I. platensis; CHSA (U15952) and CHSB (U15953) for I. triloba; CHSA (U15950) and CHSB (U15951) for I. trifida; and CHSA (U15941) and CHSB (U15942) for I. cordatotriloba. Even though the focus of this study is on the Ipomoea CHS genes, we retrieved 27 additional CHS sequences from GenBank and used them for an extensive phylogenetic analysis, including 8 petunia (Petunia hybrida) and 2 tomato (Lycopersicon esculentum) CHS genes. Note that both petunia and tomato are members of the Solanaceae family and are in the same order, Solanales, as the family Convolvulaceae, which includes Ipomoea. Those additional CHS sequences and their accession numbers are Psilotum nudum CHS (AB022682) and PnECHS (AB040027); Pinus strobus PSAJ2155 (AJ004800); Pinus sylvestris PSCHS (X60754); Pinus densiflora CHS (AB015490); Arabidopsis thaliana CHS (AF112086); Vitis vinifera VVCHS (X75969); Gerbera hybrida GHCHS1 (Z38096) and GHCHS3 (Z38098); Antirrhinum majus CHS (X03710); Scutellaria baicalensis CHS (AF035622); Perilla frutescens CHS (AB002815); Malus domestica CHS (AB074485); Glycine max GMCHS1 (X54644); Arachis hypogaea CHS1 (AY192572); Pisum sativum PSPCHS1 (X63333); Medicago sativa ALFCHS1A (L02901); Lycopersicon esculentum LETCHS1 (X55194) and LETCHS2 (X55195); and Petunia hybrida PHCHS (AF233638), PHCHSA (X14591), PHCHSB (X14592), PHCHSD (X14593), PHCHSF (X14594), PHCHSG (X14595), PHCHSJ (X14597), and PHCHSR (X04080).
The sequences were first aligned at the amino acid level using Clustal X (Thompson et al. 1997). DNA sequence alignment was then constructed according to the protein sequence alignment, followed by manual adjustment, and was used for phylogenetic and evolutionary analyses.
Phylogenetic and Evolutionary Analyses
Phylogenetic trees were reconstructed using Bayesian (Rannala and Yang 1996; Huelsenbeck and Ronquist 2001), maximum parsimony (MP), and distance-based neighbor joining (NJ; Saitou and Nei 1987) methods. The third codon positions were removed prior to phylogenetic analyses. Bayesian inference of phylogeny was carried out using MrBayes V2.01. The GTR+I+Γ model (general time-reversible with invariant sites and gamma-distributed rates for sites [Yang 1994]) was employed with the model parameters estimated from the data. Two separate runs were carried out, each with three heated chains and one cold chain. The Markov chains were run for 1,000,000 generations, and trees were sampled every 100 generations. The first 300 samples from each run were discarded as burn-in, and the remaining samples were summarized. MP and NJ analyses were performed using PAUP* 4.0 (Swofford 1998). Heuristic tree search under parsimony was conducted using the TBR (tree bisection–reconnection) swapping algorithm. NJ analysis was performed using HKY85 (Hasegawa et al. 1985) distance measure. Bootstrap analyses were conducted with 1000 replicates.
The yn00 program in the PAML package (Yang 1997; Yang and Nielsen 2000) was used to estimate synonymous and nonsynonymous substitution rates (d S and d N) between sequences. The method of Muse and Gaut (1994), implemented in Hyphy (Pond 2001), was used for codon-based relative rate tests to evaluate whether the rates of evolution at silent and replacement sites differed between the Ipomoea CHS genes. This test compares the synonymous and nonsynonymous rates between two lineages with reference to an outgroup sequence, for which we used the Antirrhinum majus CHS gene.
Maximum likelihood analysis under codon models were performed using the codeml program in the PAML package (Yang 1997). The equilibrium codon frequencies were calculated from the average nucleotide frequencies at the three codon positions. We note that due to the high sequence divergences, there is considerable variation in base compositions among sequences at the third codon positions, which may be a source of concern as the codon models used here assume a homogeneous process. See discussions below. The branch models allow the ω ratio to vary among lineages and were used to conduct likelihood ratio tests (LRT) to examine whether there was an increase in the ω ratio after gene duplications (Yang 1998; Yang and Nielsen 1998). Those models average synonymous and nonsynonymous rates over all sites in the sequence.
The site-specific models account for variable selective pressures among sites but average the synonymous and nonsynonymous rates over all lineages. We used the discrete model (M3) with K = 2 site classes (Yang et al. 2000). This model detects positive selection at individual sites only if the average d N over all lineages is higher than the average d S. If adaptive evolution occurs at a few time points and affects a few amino acids, this model might lack power in detecting positive selection. Thus the branch-site model (Model B [Yang and Nielsen 2002]) was also used, which allows the ω ratio to vary both among sites and among lineages. This model assumes four classes of sites. The first two site classes have ω0 and ω1 along all lineages in the phylogeny. The third and fourth site classes have ω0 and ω1 along all branches except a few branches of interest, which have ω2. When the estimate of ω2 is greater than 1, some sites are under positive selection along the branches of interest. This model can be compared with model M3 (K= 2), which allows for two site classes with ω0 and ω1 only, to construct a LRT. When the LRT suggests the presence of sites under positive selection, the Bayes theorem is used to calculate the posterior probabilities that each site, given the data at that site, is from the different ω classes (Nielsen and Yang 1998; Yang et al. 2000). Sites with a high probability of coming from the class with ω2 > 1 are likely to be under positive selection. Sites predicted to be under positive selection for the Ipomoea CHS genes were mapped onto the crystal structure of alfalfa CHS2 (PDB file 1BI5).
Results
Phylogeny Reconstruction
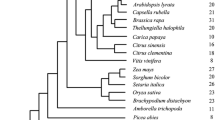
The phylogenetic relationships between Ipomoea and other CHS genes were inferred using the Bayes, MP, and NJ methods. NJ tree reconstruction used the HKY85 distance measure, and the MP tree was reconstructed using TBR perturbation algorithm. The GTR+I+Γ model of sequence evolution was used in the Bayesian analysis, which produced the tree topology in Fig. 1. The MP tree is very similar to the Bayes tree, with only minor differences concerning relationships within the clades (near the tips of the tree; see Fig. 1). The NJ tree agrees with the Bayes and MP trees concerning the groupings of the Ipomoea CHS genes and their close relatives but is different concerning the placement of the outgroup clade consisting of Malus, Glycine, Arachis, Pisum, and Medicago. The monophyly of Ipomoea CHS A, B, and C genes was strongly supported in all analyses. However, the support for the monophyly of Ipomoea CHS D and E genes is weaker. In all analyses, Ipomoea CHS A, B, and C genes clustered with the Petunia CHS B, while the Ipomoea CHS D and E clustered with other Petunia CHS genes (Fig. 1). This result is consistent with the study of Clegg and Durbin (2000). The tree topology in Fig. 1 was used in later ML analysis under models of codon substitution. We ignored uncertainties in the within-clade relationships, as previous studies demonstrated that minor differences in the tree topology had little effect (e.g., Yang et al. 2000).

Phylogeny of Ipomoea and other CHS genes inferred using the Bayesian method. P. h., Petunia hybrida. Posterior probabilities are shown for internal nodes as percentages. Branch lengths are reestimated for the Bayes tree topology under the model of codon substitution of Goldman and Yang (1994), measured by the expected number of nucleotide substitutions per codon.
Evolutionary Analysis
We calculated d S and d N in pairwise comparisons of the 18 Ipomoea CHS genes using the method of Yang and Nielsen (2000). In all 153 pairwise comparisons, we observed d N < d S. The d N values are >0.25 between subfamilies ABC and DE and <0.1 within either subfamily. We conducted the relative rate test of Muse and Gaut (1994), which tests for differences in substitution rates between two ingroup sequences relative to an outgroup sequence, for which we used Antirrhinum majus CHS. The test is applied to synonymous and nonsynonymous rates separately. The results showed significantly higher nonsynonymous rate in subfamily ABC than in subfamily DE; the smallest log-likelihood difference was 89.1, with p < 0.001 and df = 1. Significant nonsynonymous rate differences (with p < 0.05) were also detected in eight pairwise comparisons between sequences within subfamily ABC. For the synonymous rate, only 3 of the 153 comparisons showed significant rate differences. Due to the problem of multiple comparison, those few cases may well be due to chance. Those test results, combined with the estimated branch lengths on the phylogeny (Fig. 1), suggest relative homogeneous synonymous substitution rates and drastically different nonsynonymous rates between subfamilies ABC and DE.
To characterize further the variation in evolutionary rate and in selective pressure among lineages Ipomoea CHS genes, we applied two kinds of likelihood rate tests under models of codon substitution. The first analysis examines the variation of selective pressure among lineages, indicated by the nonsynonymous/synonymous rate ratio ω averaged over sites (Yang 1997, Yang 1998; Yang and Nielsen 1998). We formulate three models concerning ω ratios for branches in the tree (Table 1). The “one-ratio” model assumes the same ω ratio for all branches, and gave a log-likelihood of −22,191.06, with the estimate

= 0.077. The lower ω ratio highlights the overwhelming role of purifying selection in evolution of this gene family. The “two-ratios” model assigns two ω ratios, ω1 for branch a (ancestral to subfamily ABC and Petunia CHS B; Fig. 1) and ω0 for all other branches. The likelihood value under this model was ℓ1 = −22,190.18, with estimates

= 0.076 and

= 0.13. The small log-likelihood difference suggests no real difference in the fit of the two models.
We also fitted a “seven-ratios” model, with ω1 for branch a, ω2 for branch b, ω3 branch c, ω4 for all branches within subfamily ABC (branches after node A), ω5 for all branches within subfamily DE (branches after node B), ω6 for branches leading to Petunia CHS A, D, F, G, J, and R and tomato sequences (branches after node C), and ω0 for all other “background” branches. This model fits the data significantly better than the one-ratio model (2δℓ = 254.74, and p < 0.001 with df = 6), indicating significant variation in selective pressure among lineages. Estimates of the ω ratios under the model (Table 1) suggest that subfamily DE is under strong purifying selection, with

= 0.055 close to the background ratio
= 0.049, while branch a (with
= 0.136), branch b (with

= 0.087), branch c (with

= 0.109), and branches leading to Petunia CHS A, D, F, G, J, and R and tomato sequences (with

= 0.127) are under weaker selective constraint (Table 1). Branches within subfamily ABC clade have the highest ω ratio, with

= 0.261. The elevated ω ratios and nonsynonymous rates are compatible with both relaxed selective constraint and operation of positive selection acting on a subset of sites.
The above analysis assumes the same ω for all sites and, given the overwhelming effect of purifying selection in the CHS genes, is unlikely to identify positive selection that affects only a few sites. Thus, in the second analysis, we use the branch-site model (Model B [Yang and Nielsen 2002]) to investigate possible roles of positive selection driving functional divergence after gene duplication. When branch a is considered the foreground branch, and all other branches in the tree the background branches, parameter estimates suggested that 95.5% of sites are under selective constraint (with
= 0.027 and
= 0.189) throughout all lineages, while 4.5% of sites are under positive selection, with
= 4.94 along branch a. When branch b is considered the foreground branch, and all other branches the background branches, parameter estimates suggested that 96.4% of sites are under selective constraint (with
= 0.026 and
= 0.186) throughout all lineages, while 3.6% of sites are under positive selection with
= 52.22 along branch b. To construct LRTs, we fitted the site-specific model M3 (discrete with K = 2 site classes) as the null model, which allows only two ω ratios for all branches, estimated to be
= 0.026 and
= 0.186, and gave a log likelihood value of ℓ = −21,755.82. Comparison between the branch-site model B and the site-specific model M3 thus gave 2Δℓ = 14.8 and 2Δℓ = 11.0 for tests of branches a and b, respectively, with p < 0.005 and df = 2. Thus positive selection appears to have operated on a subset of amino acid sites along lineages a and b (Table 2). As a negative control, we also applied the branch-site model to branches within subfamily ABC clade and did not found positive selection, despite the fact that estimates of ω averaged over all sites are higher within subfamily ABC clade than along the branches ancestral to subfamily ABC clade in the branch analysis (see Table 1; seven-ratios model). Thus the higher ω ratio within subfamily ABC clade than within subfamily DE clade appears to be due to relaxed purifying selection rather than positive selection. We note that there is considerable overlap in species sampling between subfamily ABC and DE clades, so that the observed difference in the ω ratio is unlikely to be due to difference in population size or efficacy of purifying selection between the two clades.
To examine the sensitivity of the LRT to sampling of sequences included in the data set, we also analyzed a small data set consisting of only Ipomoea CHS genes, using Antirrhinum majus CHS, Scutellaria baicalensis CHS, and Perilla frutescens CHS as the outgroup. Branches a and b thus become one branch for the small data set. With the removal of distant outgroups, the sequence divergence becomes much lower, and the codon usage becomes much more homogeneous among sequences. The results obtained from this small data set (not shown) are very similar to those described above for the large data set, with some sites detected to be under positive selection along the branch separating the Ipomoea CHS ABC subfamily from the DE subfamily. Our results thus appear to be robust to minor changes to the tree topology and to sequence sampling.
To identify amino acids that might be responsible for the adaptive evolution along branches a and b, we calculated posterior probabilities that each site is from the four site classes under the branch-site model. At the p > 50% level, eight sites were identified to be under positive selection along branch a, and five sites were identified to be under positive selection along branch b. At the p > 85% level, four and two sites were identified for branches a and b, respectively. Because the empirical Bayes calculation does not account for sampling errors in the parameter estimates (Yang and Nielsen 2002) and because the inference is performed on every site, the results have to be taken with caution. For comparison, we also performed ML reconstruction of ancestral sequences using the codon model of Goldman and Yang (1994). The reconstruction suggested 67 amino acid replacements along branch a and 61 amino acid replacements along branch b. All sites, except for one, that are inferred to be under positive selection under the branch-site model are on the list of changes based on ancestral reconstruction.
The crystal structure of chalcone synthase CHS2 from the alfalfa (Medicago sativa) has been determined by Ferrer et al. (1999). The structure revealed that four chemically reactive residues (Cys164, Phe215, His303, and Asn336), which are conserved in all the known CHS-related enzyme, define the active site, five residues (Ser133, Glu192, Thr194, Thr197, and Ser338) form the coumaroyl-binding pocket, and seven residues (Thr132, Met137, Phe215, Ile254, Gly256, Phe265, and Pro375) form the cyclization pocket. Moreover, all CHS-like proteins exhibit strong conservation of residues shaping the geometry of the active site (Pro138, Gly163, Gly167, Leu214, Asp217, Gly262, Pro304, Gly305, Gly306, Gly335, Gly374, Pro375, and Gly376) (Ferrer et al. 1999).
Comparison of the protein sequences of alfalfa CHS2 and the Ipomoea CHS indicated that the Ipomoea sequences of subfamily DE possess exactly the same amino acids as alfalfa CHS2 at the active site, the coumaroyl-binding pocket, and the cyclization pocket. Sequences of subfamily ABC have conserved residues at the active site as well, but some sites (Thr194, Thr197, Ile254, Gly256, and Ser338) involved in the formation of the coumaroyl-binding pocket and the cyclization pocket experienced amino acid replacements. The likelihood analysis suggested that one of those replacements (Gly256) happened along branch a and was driven by positive selection with the posterior probability p = 0.98.
The likelihood analysis also suggested some other sites not directly involved in the formation of the initiation/elongation/cyclization cavity to be under positive selection. Interestingly, those sites are scattered over the entire primary sequence but, when mapped onto the crystal structure (Fig. 2), tend to be clustered around the active site, suggesting that they might also have affected the configuration and function at the active site.

Location of amino acid residuals identified as likely to be under positive selection in Ipomoea CHS genes using the structure of alfalfa CHS2 (PDB file 1BI5) as a template. Theactive sites are shown in black spacefill. The residuals identified as under positive selection are in gray spacefill, and most of them are around the active sites.
Discussion
Gene duplication is often followed by accelerated evolution (Li 1985; Bielawski and Yang 2001), which can be due to either positive Darwinian selection for functional divergence (Ohta 1993) or relaxation of selective constraints (Kimura 1983). In the former case, the requirement of new function exerts directional selective pressure, driving the fixation of advantageous nonsynonymous mutations. In the latter case, neutral mutations are fixed at random, which, perhaps due to environmental changes, lead to a novel function in one or both copies.
Many plant species are found to contain small multigene families of CHS genes (Koes et al. 1989; Durbin et al. 1995; Helariutta et al. 1996). Analysis of CHS multigene family suggested recurrent gene duplications and subsequent adaptive differentiation among duplicated copies (Durbin et al. 2000; Helariutta et al. 1996). Some other plant-specific polyketide synthases, including STS, ACS, BBS, 2PS, and PVPS, are proposed to have evolved from CHSs by the same mechanism (Schröder 1997; Lukacin et al. 1999; Preisig-Müller et al. 1995; Eckermann et al. 1998; Paniego et al. 1999). Those enzymes share a common chemical mechanism with CHS but differ from CHS in their substrate specificity and/or in the stereochemistry of the polyketide cyclization reaction. Because of the similarity both in the reactions catalyzed and in sequences, it was proposed that these enzymes formed a family called the CHS superfamily (Schröder 1997). As many as 150 CHS-related sequences have been cloned from various plants (Ferrer et al. 1999). However, the functional diversity of CHS-related proteins is not yet fully explored. Schröder (1997) pointed out that too many chalcone synthase entries in public sequence databases are solely identified by sequence similarity. It is likely that some of the sequences putatively identified as chalcone synthase on the basis of their sequence similarity actually encode related enzymes. The clone of CHS2 from Gerbera hybrida is an example. It was originally annotated as a CHS. Later, Eckermann et al. (1998) convincingly showed that it serves an alternate function in the biosynthesis of pyrone glucosides that contribute to insect and pathogen resistance in Gerbera hybrida. It was thus renamed 2-pyrone synthase (2PS). Based on the similarity of 2PS to the CHSs in Gerbera hybrida and the sporadic distribution of gerberin-type compounds, it was proposed that 2PS evolved from CHS by gene duplication and subsequent differentiation, and positive selection was believed to drive the differentiation among duplicated CHS copies (Yang et al. 2002).
Five functional CHS genes have been reported in the morning glories. Currently all these sequences are annotated as CHS sequences. However, the enzyme functions of these genes have not yet been clearly demonstrated. The substrate and product specificities of these sequences remain unknown. The relative rate tests and the codon-based likelihood analysis in this study suggested that the nonsynonymous rate is higher in subfamily ABC than in subfamily DE. Maximum-likelihood analysis under the branch models also indicated the role of positive selection along the branch leading to subfamily ABC, even though most of sites in the gene are under strong purifying selection. This result, together with the expression patterns of CHS D and E in morning glories (Durbin et al. 2000), leads us to conclude that some amino acid replacements along the branch ancestral to subfamily ABC are driven by Darwinian selection driving functional divergence.
Of the five amino acid replacements that are involved in the formation of the coumaroyl-binding pocket and the cyclization pocket experienced amino acid replacements along the branches ancestral to subfamily ABC, one (G256L) was identified to be under positive selection by the likelihood analysis. Site Gly256 is involved in the formation of the cyclization pocket and is conserved in all CHSs, STSs, ACSs, and BBSs. However, natural variation at this position occurs in Gerbera hybrida 2PS, Petunia CHS B, and Ipomoea CHS A, B, and C (Jez et al. 2001), with glycine replaced by leucine. Jez et al. (2000, 2001) constructed a CHS G256L mutant from the alfalfa CHS2 gene by site-directed mutagenesis. Comparison of the molecular surface of the initiation/elongation/cyclization cavity of wild-type CHS and the G256L mutant revealed that in the mutant, the cavity volume is reduced from 605 to 572 Å3. Functionally, the mutant is involved in triketide formation, and the wild type in tetraketide synthesis. The mutant thus produces methylpyrone and bis-noryangonin, instead of naringenin chalcone, from acetyl-CoA and malonyl-CoAs. Similarly, biochemical analysis of natural Ipomoea CHS genes revealed that only CHS D and E genes are capable of catalyzing the condensation reaction that results in naringenin chalcone, while the CHS A and B genes appear to encode enzymes that produce bis-noryangonin but not naringenin chalcone (Clegg and Durbin 2000). Therefore, sequence divergences between Ipomoea CHS A, B, and C and CHS D and E genes correlate with functional changes in the kinetic and specificity properties. Positive Darwinian selection seem to have promoted the divergence of subfamily ABC and subfamily DE and is at least partially responsible for a rate increase following gene duplication.
The Petunia CHS gene family had been extensively characterized, with as many as 12 genes provisionally identified (Koes et al. 1987). Eight complete CHS genes of Petunia were used in this study. The Petunia CHS genes appear to share common lines of descent with the Ipomoea CHS genes, with the majority of Petunia CHS genes close to Ipomoea CHS D and E, and Petunia CHS B close to Ipomoea CHS A, B, and C. This pattern seems to suggest an ancient gene duplication prior to the divergence between Solanaceae and Convolvulaceae. Durbin et al. (2000) estimated that the divergence between Ipomoea subfamilies ABC and DE occurred more than 100 Myr ago, while the time of divergence between Solanaceae and Convolvulaceae is about 70 Myr ago. The long branch for Petunia CHS B and Ipomoea CHS A, B, and C in Fig. 1 indicates an accelerated evolution after gene duplication, which led to the substantial sequence divergence between subfamily ABC and DE in Ipomoea and Petunia CHS B and the remaining Petunia CHS sequences.
Currently, the phenotypic effects of the CHS variants in Ipomoea and Petunia remain to be established. Comparative analysis of gene expression patterns in Ipomoea showed that CHS D is evidently responsible for the accumulation of pigment in Ipomoea, whereas CHS A, B, and C are mainly expressed in the unpigmented tube and expressed at a low level in floral limb. It is unclear, so far, whether mutations of CHS A, B, and C genes have a direct impact on divergent pigmentation patterns in Ipomoea. Generally loss of CHS function results in a lack of anthocyanin and an albino flower color phenotype. This is not unexpected considering that CHS is a key enzyme in the anthocyanin pathway. However, it is now known that CHS is encoded by a small multigene family in many species including those species containing mutations that result in loss of CHS activity (Durbin et al. 2000). If there is redundancy in function, then presumably another CHS gene family member would assume the function of a lost CHS member and there would be no loss of pigmentation observed in mutant phenotypes (Durbin et al. 2000). Genetic redundancy must be of some adaptive value to the plant to persist over evolutionary time. Without positive selection for the maintenance of sequence fidelity, redundant gene copies will erode over time due to accumulation of deleterious mutations (Walsh 1995). Natural variation in Ipomoea CHS A, B, and C genes does not result in functionally impaired enzymes but, in fact, generates catalytically active enzymes that display altered substrate and product specificities. It is therefore a prime facie evidence that the duplicate is positively selected. The evolution of new catalytic functions from CHS genes is also found elsewhere, for example, at least three independent shifts from CHS to stilbene synthase (STS) have arisen in seed plant evolution (Tropf et al. 1994). Catalytic shifts from CHS to acridone synthase (ACS) (Lukacin et al. 1999), bibenzyl synthase (BBS) (Preisig-Müller et al. 1995), 2-pyrone synthase (2PS) (Eckermann et al. 1998), and phlorisovalerophenone synthase (PVPS) (Paniego et al. 1999) are also evident. Therefore, gene duplication, coupled with functional divergence, is a recurrent pattern in the evolution of the CHS gene family (Clegg and Durbin 2003).
The likelihood analysis showed that along branches a and b, some sites not directly involved in the formation of the initiation/elongation/cyclization cavity appear to be under positive selection as well. Most of these sites are clustered around the active site in the crystal structure (Fig. 2). It is not yet established whether changes at these sites are involved in a shift in enzymatic function. However, Staffard (1991) postulated that the flavonoid pathway might be organized into aggregates or complexes such as exist in other pathways (Hrazdina and Jensen 1992; Srere et al. 1987; Srivastava and Bernhard 1986). Such complexes offer many advantages in terms of kinetics, channeling of intermediates, and protection of labile intermediates (Debnam et al. 1997). Srere et al. (1987) postulated that if a protein were to function as part of a metabolon, it must have conserved binding sites for maintenance of the complex and that it may be these binding sites that distinguish different isozymes. It is possible that the amino acid differences observed in the Ipomoea CHS genes could be involved in directing the protein into the correct complex (Durbin et al. 2000).
References
JP Bielawski Z Yang (2001) ArticleTitlePositive and negative selection in the DAZ gene family. Mol Biol Evol 18 523–529 Occurrence Handle1:CAS:528:DC%2BD3MXis1eisLk%3D Occurrence Handle11264403
MT Clegg ML Durbin (2000) ArticleTitleFlower color variation: A model for the experimental study of evolution. Proc Natl Acad Sci USA 97 7016–7023 Occurrence Handle10.1073/pnas.97.13.7016 Occurrence Handle1:CAS:528:DC%2BD3cXksVKisb0%3D Occurrence Handle10860965
MT Clegg ML Durbin (2003) ArticleTitleTracing floral adaptations from ecology to molecules. Nature Rev Genet 4 260–215
PM Debnam G Shearer L Blackwood DH Kohl (1997) ArticleTitleEvidence for channeling of intermediates in the oxidative pentose phosphate pathway by soybean and pea nodule extracts, yeast extracts, and purified yeast enzymes. Eur J Biochem 246 283–290 Occurrence Handle1:CAS:528:DyaK2sXjvVGrtrk%3D Occurrence Handle9208916
ML Durbin GH Learn GA Huttley MT Clegg (1995) ArticleTitleEvolution of the chalcone synthase gene family in the genus Ipomoea. Proc Natl Acad Sci USA 92 3338–3342 Occurrence Handle1:CAS:528:DyaK2MXltFSntbc%3D Occurrence Handle7724563
ML Durbin B McCaig MT Clegg (2000) ArticleTitleMolecular evolution of the chalcone synthase multigene family in the morning glory genome. Plant Mol Biol 42 79–92 Occurrence Handle10.1023/A:1006375904820 Occurrence Handle1:CAS:528:DC%2BD3cXht1Gku7c%3D Occurrence Handle10688131
S Eckermann G Schröder J Schmidt D Strack RA Edrada Y Helariutta P Elomaa I Kotilainen P Proksch TH Teeri J Schröder (1998) ArticleTitleNew pathway to polyketides in plants. Nature 396 390–397 Occurrence Handle10.1038/24652 Occurrence Handle9845078
JL Ferrer JM Jez ME Bowman RA Dixon JP Noel (1999) ArticleTitleStructure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nature Struct Biol 6 775–784 Occurrence Handle10.1038/11553 Occurrence Handle1:CAS:528:DyaK1MXltVShsLg%3D
S Fukada-Tanaka A Hoshino Y Hisatomi Y Habu M Hasebe S Iida (1997) ArticleTitleIdentification of new chalcone synthase genes for flower pigmentation in the Japanese and common morning glories. Plant Cell Physiol 38 754–758 Occurrence Handle1:CAS:528:DyaK2sXktVKkur4%3D Occurrence Handle9249990
N Goldman Z Yang (1994) ArticleTitleA codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11 725–736 Occurrence Handle7968486
M Hasegawa H Kishino T Yano (1985) ArticleTitleDating the human-ape split by a molecular clock of mitochondrial DNA. J Mol Evol 22 160–174 Occurrence Handle1:CAS:528:DyaL2MXmtFSns7g%3D Occurrence Handle3934395
Y Helariutta M Kotilainen P Elomaa N Kalkkinen K Bremer TH Teeri VA Albert (1996) ArticleTitleDuplication and functional divergence in the chalcone synthase gene family of Asteraceae: Evolution with substrate change and catalytic simplification. Proc Natl Acad Sci USA 93 9033–9038 Occurrence Handle10.1073/pnas.93.17.9033 Occurrence Handle1:CAS:528:DyaK28Xlt1Chsrw%3D Occurrence Handle8799149
G Hrazdina RA Jensen (1992) ArticleTitleSpatial organization of enzymes in plant metabolic pathways. Annu Rev Plant Physiol Plant Mol Biol 43 241–267 Occurrence Handle10.1146/annurev.pp.43.060192.001325 Occurrence Handle1:CAS:528:DyaK38Xks1Knur0%3D
JP Huelsenbeck F Ronquist (2001) ArticleTitleMrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17 754–755 Occurrence Handle10.1093/bioinformatics/17.8.754 Occurrence Handle1:STN:280:DC%2BD3MvotV2isw%3D%3D Occurrence Handle11524383
JM Jez MB Austin JL Ferrer ME Bowman J Schröder JP Noel (2000) ArticleTitleStructural control of polyketide formation in plant-specific polyketide synthases. Chem Biol 7 919–930 Occurrence Handle1:CAS:528:DC%2BD3MXltFegtg%3D%3D Occurrence Handle11137815
JM Jez ME Bowman JP Noel (2001) ArticleTitleStructure-guided programming of polyketide chain-length determination in chalcone synthase. Biochemistry 40 14829–14838 Occurrence Handle10.1021/bi015621z Occurrence Handle1:CAS:528:DC%2BD3MXotFyrsrY%3D Occurrence Handle11732902
M Kimura (1983) The neutral theory of molecular evolution. Cambridge University Press Cambridge, UK
RE Koes CE Spelt JNM Mol AG Gerats (1987) ArticleTitleThe chalcone synthase multigene family of Petunia hybrida (V30): Sequence homology, chromosomal localization and evolutionary aspects. Plant Mol Biol 10 159–169 Occurrence Handle1:CAS:528:DyaL1cXovVymuw%3D%3D
RE Koes CE Spelt PJM van den Elzen JNM Mol (1989) ArticleTitleCloning and molecular characterization of the chalcone synthase multigene family of Petunia hybrida. Gene 81 245–257 Occurrence Handle10.1016/0378-1119(89)90185-6 Occurrence Handle1:CAS:528:DyaK3cXitFWksLc%3D Occurrence Handle2806915
W-H Li (1985) Accelerated evolution following gene duplication and its implications for the neutralist-selectionist controversy. T Otha K Aoki (Eds) Population genetics and molecular evolution. Japan Scientific Press, Tokyo 333–352
W-H Li (1997) Molecular evolution. Sinauer Sunderland, MA
R Lukacin K Springob C Urbanke C Ernwein G Schröder J Schröder U Matern (1999) ArticleTitleNative acridone synthase I and II from Ruta graveolens L. form homodimers. FEBS Lett 448 135–140 Occurrence Handle10.1016/S0014-5793(99)00355-5 Occurrence Handle1:CAS:528:DyaK1MXit1Wrtbw%3D Occurrence Handle10217426
SV Muse BS Gaut (1994) ArticleTitleA likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates with application to the chloroplast genome. Mol Bio Evol 11 715–724 Occurrence Handle1:CAS:528:DyaK2cXlvFOjsL8%3D
R Nielsen Z Yang (1998) ArticleTitleLikelihood models for detecting positively selected amino acid sites and application to the HIV-1 envelop gene. Genetics 148 929–936 Occurrence Handle1:CAS:528:DyaK1cXks1eitr8%3D Occurrence Handle9539414
S Ohno (1970) Evolution by gene duplication. Springer-Verlag New York
T Ohta (1993) ArticleTitlePattern of nucleotide substitution in growth hormone-prolactin gene family: A paradigm for evolution by gene duplication. Genetics 134 1271–1276 Occurrence Handle1:CAS:528:DyaK2cXht1KltLs%3D Occurrence Handle8375661
NB Paniego KWM Zuurbier SY Fung R Van der Heijden JJC Scheffer R Verpoorte (1999) ArticleTitlePhlorisovalerophenone synthase, a novel polyketide synthase from hop (Humulus lupulus L.) cones. Eur J Biochem 262 612–616 Occurrence Handle10.1046/j.1432-1327.1999.00444.x Occurrence Handle1:CAS:528:DyaK1MXjvVGmtLg%3D Occurrence Handle10336650
SK Pond (2001) Hypothesis testing using phylogenies (HYPHY), version 0.91 beta. University of Arizona Tucson
R Preisig-Müller P Gnau H Kindl (1995) ArticleTitleThe inducible 9,10-dihydrophenanthrene pathway: Characterization and expression of bibenzyl synthase and S-adenosylhomocysteine hydrolase. Arch Biochem Biophys 317 201–207 Occurrence Handle10.1006/abbi.1995.1154 Occurrence Handle7872785
B Rannala Z Yang (1996) ArticleTitleProbability distribution of molecular evolutionary trees: A new method of phylogenetic inference. J Mol Evol 43 304–311 Occurrence Handle1:CAS:528:DyaK28XlvVahur8%3D Occurrence Handle8703097
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
A Schöppner H Kindl (1984) ArticleTitlePurification and properties of a stilbene synthase from induced cell suspension cultures of peanut. J Biol Chem 259 6806–6811 Occurrence Handle1:CAS:528:DyaL2cXkvVKit7g%3D Occurrence Handle6427224
J Schröder (1997) ArticleTitleA family of plant-specific polyketide synthases: Facts and predictions. Trends Plant Sci 2 373–378 Occurrence Handle10.1016/S1360-1385(97)01104-7
PA Srere B Sumegi AD Sherry (1987) ArticleTitleOrganizational aspects of the citric acid cycle. Biochem Soc Symp 54 173–182 Occurrence Handle1:CAS:528:DyaL1cXmt1Cru74%3D Occurrence Handle3332994
DK Srivastava SA Bernhard (1986) ArticleTitleMetabolite transfer via enzyme-enzyme complexes. Science 234 1081–1084 Occurrence Handle1:CAS:528:DyaL2sXotVCitw%3D%3D Occurrence Handle3775377
HA Stafford (1991) ArticleTitleFlavonoid evolution: An enzymic approach. Plant Physiol 96 680–685 Occurrence Handle1:CAS:528:DyaK3MXltFSmsbc%3D
DL Swofford (1998) PAUP*: Phylogenetic analysis using parsimony (* and other methods), version 4.0. Sunderland, MA
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe CLUSTAL-X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25 4876–4882 Occurrence Handle1:CAS:528:DyaK1cXntFyntQ%3D%3D Occurrence Handle9396791
S Tropf T Lanz SA Rensing J Schröder G Schröder (1994) ArticleTitleEvidence that stibene synthases have developed from chalcone synthases several times in the course of evolution. J Mol Evol 38 610–618 Occurrence Handle1:CAS:528:DyaK2cXkt12rt70%3D Occurrence Handle8083886
JB Walsh (1995) ArticleTitleHow often do duplicated genes evolve new functions? Genetics 139 421–428 Occurrence Handle1:STN:280:ByqB38%2FmvVQ%3D Occurrence Handle7705642
J Yang J Huang H Gu Y Zhong Z Yang (2002) ArticleTitleDuplication and adaptive evolution of the chalcone synthase genes of Dendranthema (Asteraceae). Mol Biol Evol 19 1752–1759 Occurrence Handle1:CAS:528:DC%2BD38XnvFSlu7w%3D Occurrence Handle12270901
Z Yang (1994) ArticleTitleMaximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39 306–314 Occurrence Handle7932792
Z Yang (1997) ArticleTitlePAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13 555–556 Occurrence Handle9367129
Z Yang (1998) ArticleTitleLikelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15 568–573 Occurrence Handle9580986
Z Yang JP Bielawski (2000) ArticleTitleStatistical methods for detecting molecular adaptation. Trends Ecol Evol 15 496–503 Occurrence Handle10.1016/S0169-5347(00)01994-7 Occurrence Handle11114436
Z Yang R Nielsen (1998) ArticleTitleSynonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol 46 409–418 Occurrence Handle1:CAS:528:DyaK1cXitlSgu7w%3D Occurrence Handle9541535
Z Yang R Nielsen (2000) ArticleTitleEstimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17 32–43 Occurrence Handle1:CAS:528:DC%2BD3cXotF2qtA%3D%3D Occurrence Handle10666704
Z Yang R Nielsen (2002) ArticleTitleCodon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol 19 908–917 Occurrence Handle1:CAS:528:DC%2BD38Xks1Ojtbk%3D Occurrence Handle12032247
Z Yang R Nielsen N Goldman AMK Pedersen (2000) ArticleTitleCodon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155 431–449
Acknowledgements
We thank two anonymous reviewers for comments. This study was supported by National Natural Science Foundation of China Grant 39830020 to H.G., and HFSP Grant Y0055/2001-M and BBSRC Grant 31/G14969 to Z.Y.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, J., Gu, H. & Yang, Z. Likelihood Analysis of the Chalcone Synthase Genes Suggests the Role of Positive Selection in Morning Glories (Ipomoea) . J Mol Evol 58, 54–63 (2004). https://doi.org/10.1007/s00239-003-2525-3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2525-3