
Abstract
The majority of known plant resistance genes encode proteins with conserved nucleotide-binding sites and leucine-rich repeats (NBS-LRR). Degenerate primers based on conserved NBS-LRR motifs were used to amplify analogues of resistance genes from the dicot sugar beet. Along with a cDNA library screen, the PCR screen identified 27 genomic and 12 expressed NBS-LRR RGAs (nlRGAs) sugar beet clones. The clones were classified into three subfamilies based on nucleotide sequence identity. Sequence analyses suggested that point mutations, such as nucleotide substitutions and insertion/deletions, are probably the primary source of diversity of sugar beet nlRGAs. A phylogenetic analysis revealed an ancestral relationship among sugar beet nlRGAs and resistance genes from various angiosperm species. One group appeared to share the same common ancestor as Prf, Rx, RPP8, and Mi, whereas the second group originated from the ancestral gene from which 12C1, Xa1, and Cre3 arose. The predicted protein products of the nlRGAs isolated in this study are all members of the non-TIR-type resistance gene subfamily and share strong sequence and structural similarities with non-TIR-type resistance proteins. No representatives of the TIR-type RGAs were detected either by PCR amplification using TIR type-specific primers or by in silico screening of more than 16,000 sugar beet ESTs. These findings suggest that TIR type of RGAs is absent from the sugar beet genome. The possible evolutionary loss of TIR type RGAs in the sugar beet is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genes conferring gene-for-gene disease resistance in plants (R-genes) can be categorized into distinct classes based on the predicted protein structures (reviewed by Baker et al. 1997; Dangl and Jones 2001). The majority of cloned R-genes fall into the nucleotide-binding site/leucine-rich repeat (NBS-LRR)-containing gene family. Members of this family have been identified in both dicots and monocots and confer resistance to a variety of plant pathogens, including bacteria, fungi, viruses, and nematodes (Hammond-Kosack and Jones 1997; Wang et al. 1999). The NBS-LRR disease resistance genes are thought to function in both elicitor recognition and activation of downstream signal pathways leading to disease resistance responses (Jones and Jones 1997; van der Biezen et al. 2000).
The NBS-LRR resistance genes are characterized by several domains. These domains include a variable N-terminal domain of approximately 200 amino acids, a putative nucleotide-binding site (NBS) of approximately 300 amino acids, and a C-terminal tandem array of approximately 10–40 short LRR motifs. The central NBS domain, which consists of P-loop/kinase-1a, kinase-2, and kinase-3a motifs, is highly conserved in diverse organisms in a wide variety of ATP- or GTP-binding proteins (Traut 1994). This domain shares high homology with the Apaf-1 and Ced-4 genes that are involved in animal innate immunity and apoptosis (van der Biezen and Jones 1998; Aravind et al. 1999), implicating a conserved regulatory mechanism of cell death programmes in plants, mammals, and other animals. The NBS domain is therefore referred to as NB-ARC (Apaf-1, R-protein and Ced-4) domain. Elicitor recognition specificity is believed to be determined by the LRR domain (Ellis et al. 1999). The LRR domain is the most variable region among related proteins, presumed to have diverged throughout evolution, and ensures recognition-dependent activation of resistance (Shen et al. 1998; Noel et al. 1999). The N-terminus, probably together with the NBS-domain, is thought to participate in activating downstream signal transduction components (Aarts et al. 1998a; van der Biezen et al. 2000). Taking the N-terminal domain structure into account, the NBS-LRR class of resistance genes can be divided into two subfamilies: TIR and CC/non-TIR (Meyers et al. 1999; Pan et al. 2000a). TIR type proteins share homology with the Drosophila Toll and human Interleukin receptor-like regions, while the non-TIR subfamily generally has a coiled-coil (CC) or leucine zipper structure. It is tempting to speculate that the analogous roles of the N-termini of the TIR and CC/non-TIR resistance proteins are to recruit specifically other effectors through protein–protein interactions (Horng et al. 2001; Jebanathirajah et al. 2002).
To date, more than 30 functionally defined NBS-LRR resistance genes and a large number of resistance gene homologues from diverse taxa are available in databases. PCR amplification using primers based on conserved motifs, particularly within the NBS domain, has been used successfully to isolate NBS-LRR resistance gene analogues from a wide variety of plant species (Aarts et al. 1998a; Leister et al. 1998; Rivkin et al. 1999; Speulman et al. 1998; Shen et al. 1998; Timmerman-Vaughan et al. 2000). These sequences, together with genomic sequences and ESTs, offer an opportunity to evaluate the dynamics and speciation of NBS-LRR resistance gene evolution (Meyers et al. 1999; Pan et al. 2000a; Cannon et al. 2002). Recent research supports an ancient origin and divergent evolution of TIR and non-TIR resistance gene subfamilies in cereals and dicots. TIR type resistance genes are not found in current cereal public databases, and all RGAs isolated from cereal are clustered into the non-TIR clade, suggesting a complete loss of TIR type resistance genes from cereal genomes (Pan et al. 2000a). Members of both TIR and non-TIR types of disease resistance genes, as well as RGAs, are widely distributed among dicot species (Cannon et al. 2002).
Sugar beet (Beta vulgaris L.) belongs to the family Chenopodiaceae. Due to its agricultural importance and relatively small genome, of 758 Mbp (Arumuganathan and Earle 1991), sugar beet is a model plant for root-storing species. Several genes for disease resistance have been cloned or mapped with molecular markers. ESTs are available in public databases, the majority of which stem from a normalized cDNA library created by the oligonucleotide fingerprinting technique (Herwig et al. 2002). Recently, several RGAs have been amplified from genomic DNA and cDNAs of sugar beet (Hunger et al. 2003). Here, we report the isolation and phylogenetic analysis of NBS-LRR RGAs (nlRGAs) from sugar beet and demonstrate that the sugar beet nlRGAs analyzed are all members of the non-TIR resistance gene subfamily.
Materials and Methods
Oligonucleotide Primers and PCR Amplification
Five amino acid motifs of the NBS domain conserved among known resistance proteins were chosen for the design of oligonucleotide primers to amplify nlRGAs from sugar beet genomic DNA (Table 1). The motifs are P-loop/kinase-1a, kinase-2, kinase-3a, hydrophobic domain (GLPL), and TIR/non-TIR consensus sequences. Five degenerate primers were designed in the sense direction according to the predicted amino acid sequences of the P-loop, kinase-2, and kinase-3a domains, and three antisense primers corresponding to the kinase-3a and GLPL domains were designed. Two sets of gene-specific primers were designed based on the P-loop and GLPL domains of the nematode resistance genes Mi and Gpa2. Two sets of degenerate primers specific for TIR and four for non-TIR NBS-LRR-containing resistance genes were designed from the TIR- and non-TIR-type consensus sequences described by Cannon et al. (2002). In total, 24 primer combinations were used for PCR amplification.
PCR Amplification, DNA Fragment Cloning, and Sequence Analysis
The nematode (Heterodera schachtii Schm.) resistant sugar beet line A906001 (kindly provided by A. Dieckmann-Heimburg, Nienstädt, Germany) were grown in a greenhouse at 25°C. Genomic DNA was extracted from sugar beet leaves as described by Rogers and Bendich (1985). Genomic DNA (200 ng) from A906001 was used as a PCR template. PCR was performed in the presence of 50 pmol forward and reverse primers, 1 unit Taq polymerase (Invitrogen, Karlsruhe, Germany), 0.5 mM dNTPs, 2 mM MgCl2, pH 8.3, in 50 µl reaction volumes. The optimal annealing temperature for each primer combination was determined by a gradient PCR reaction using a T-gradient thermal cycler (Biometra, Göttingen, Germany), and single-primer reactions were used as controls. DNA was denatured at 94°C for 3 min, followed by 35 cycles of 1 min at 94°C, 1 min at 40°C–60°C (depending on primer combinations), and 1.5 min at 72°C. PCR products were separated on 1.5% agarose gels, and fragments of expected sizes (250–600 bp) were extracted and purified using a gel extraction kit (Qiagen, Hilden, Germany). Fragments were cloned using the pGEM-T vector (Promega, Madison, WI). Plasmid DNA of the clones was isolated using a GFX micro plasmid prep kit (Amersham Pharmacia Biotech, Freiburg, Germany). The plasmid inserts were analyzed by double digestion with ApaI and PstI. Sequencing of the inserts was performed using a Li-Cor-sequencer 4000, following the protocol of Sanger (1977), using IRD800-labeled M13 forward and reverse primers (MWG, Ebersberg, Germany) and the SequiTherm EXEL II Long-Read DNA Sequencing Kit LC (EPICENTRE Technologies, Madison, WI). Sequences were analyzed using LaserGene software (DNASTAR, Madison WI).
cDNA Library Screening and Amplification of cDNA Ends
A λ-ZapII cDNA library, made from roots of the sugar beet line A906001, was screened with the genomic DNA fragment gZR-3, amplified using primers R-1 and R-6 (Table 2), according to the manufacturer’s instructions (Stratagene, CA). Phage-blots were hybridized with 32P-labeled DNA probes at 45, 50, 55, and 60°C, washed twice (0.5 × SSC, 0.2% [w/v] SDS) for 30 min, and exposed at −70°C for 48 h. Forty positive plaques were obtained in the first screening. After the second and third rounds, 25 individual plaques were recovered and used for excision in vivo as described by the manufacturer (Stratagene). For each positive plaque, four independent plasmid clones were sequenced.
Full-length cDNAs were cloned using a SMART RACE kit (Clontech, Heidelberg, Germany) following the manufacturer’s instructions. First-strand cDNA was synthesized from 1 µg poly(A)+ RNA isolated from roots of the sugar beet line A906001. Primary RACE products were confirmed by a second round of PCR using nested gene-specific primers. Positive RACE products were isolated from agarose gels, subcloned into the pGEM-T vector, and sequenced as described above.
Multiple Sequence Alignment, Sequence Similarity Search, and Phylogenetic Tree Construction
RGAs and known NBS-LRR-type resistance genes from GenBank were aligned using ClustalW (Thompson et al. 1994) on the European Bioinformatics Institute (EBI) server. Similarity searches against GenBank (NCBI, Release 130.0) and databases were done using TBLASTN (Altschul et al. 1997) with NBS and TIR consensus sequences as queries. Two NBS-type consensus sequences (TIR NBS-HMM and non-TIR NBS-HMM) were generated by the hidden Markov model (HMM) with NBS_SubDomA and B profiles (http://niblrrs.ucdavis.edu/ [see supplementary material 1]). The TIR consensus sequence was generated based on an alignment of TIR type resistance genes in Group I (Fig. 2). HMM-based and SAM-T99 database (Park et al. 1998) searches were performed on 12 September 2002. Consensus amino acids at relative positions were calculated based on multiple alignments of 876 returned sequences (hits) with cutoffs of less than 0.01. A phylogenetic tree was created using the 400 amino acids spanning the NBS domains from a full-length protein sequence alignment (created with ClustalW at EBI) using the neighbor-joining method with ClustalW at the Pasteur Institute (settings included Kamura’s correction and Ignore positions with gaps, bootstrap values of 1000, and two seed numbers). The tree was drawn using TreeView (Page et al. 1996).

Multiple sequence alignment analysis of the NBS domains of four representative sugar beet nlRGAs in comparison with 20 known disease resistance genes. Conserved residues are shaded by threeshade levels, which indicated conserved percentages of >90, >70, and >50% from darkest to lightest. Motifs are defined as in the Pfam hidden Markov model for NB-ARC sequences (http://pfam.wustl.edu/cgi-bin/getdesc?name=NB-ARC ; Fourmann et al. 2001). Delineation of Groups I and II (corresponding to TIR and non-TIR disease resistance genes, respectively) was made according to Pan et al. (2000b).
Determining Compositional Gradients Within Genes
Compositional gradients were detected using SAGS (Software of Analyzing Gene Structure; www.cab.zju.edu.cn/instzzkx/laborate/Bioinplant/bioinplant_page.htm). The Codon Usage Database (http://www.kazusa.or.jp/codon/ ) was used for pattern analysis of GC content in the three codon positions and for codon usage in general. At least 74 coding sequences (CDS) were downloaded from each of five cereals (Oryza sativa, Zea may, Hordeum vulgare, Sorghum bicolor, and Triticum aestivum) and seven dicot species (Arabidopsis thaliana, Medicago sativa, Glycine max, Lycopersicon esculentum, Brassica napus, Spinacia oleracea, and Beta vulgaris) (Table 5). For codon usage analysis of R-genes or nlRGAs, four full-length nlRGAs from sugar beet [cZR-1(f), cZR-3(f), cZR-7(f), and cZR-9(f)], four disease resistance genes from rice (Pib, BAA76282; Xa21, AAC49123; Xa1, BAA25068; Pi-ta, AAK00132), and five from Arabidopsis (RPM1, A57072; RPP13, T51186; RPS2, A54809; RPS5, AAC26126; RPP1, AAC72979) were compiled into a codon usage table using the count codon program at the Codon Usage Database website. Twenty-five full-length sugar beet protein sequences (including methionine starts and more than 60 amino acids) from the SWISS-PROT database were randomly selected for determining the frequency of serines and alanines using the method described by Wong et al. (2002).
Results
Isolation of Genomic and Expressed nlRGAs from Sugar Beet
PCR amplification on sugar beet genomic DNA using each degenerate primer combination (Tables 1 and 2) yielded products of various sizes. Fragments between 250 and 650 bp in size amplified with P-loop/kinase-3a, P-loop/GLPL, kinase-2/GLPL, TIR-type, and non-TIR-type specific primer combinations were cloned and sequenced. Of 215 cloned fragments, 65 shared sequence homology with NBS-LRR resistance genes (data not shown). Of these clones, 27 had potential open reading frames encoding at least one internal motif in addition to the two primer-targeted motifs of the NBS domain (Table 2). They were therefore designated sugar beet genomic nlRGAs gZR-1 to gZR-27. Unexpectedly, no nlRGAs were obtained using the four TIR type-specific primers.
Expressed RGAs are considered to be potentially functional genes that may amplify or directly activate resistance responses. To isolate expressed nlRGAs, the genomic sequence of gZR-3, which shows the highest homology at the nucleotide level with both TIR and non-TIR types of known resistance genes, was used as a probe to screen a sugar beet cDNA library under various stringent conditions. In this screen, 12 distinct nlRGAs, designated cZR-1 to cZR-12, were identified. Five of these were novel, and seven corresponded to the genomic nlRGAs cloned. No introns were detected in the genomic nlRGAs (data not shown).
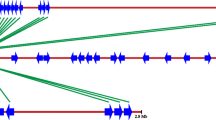
On the basis of nucleotide sequence identity, 32 nlRGAs (20 genomic and 12 cDNAs) were classified into three subfamilies (Table 2). Sequences within a subfamily share greater than 90% nucleotide sequence identity; different subfamilies share less than 65% similarity (Fig. 1A and B). Sequence analysis using the consensus region of each subfamily revealed numerous mutations, including nucleotide substitutions and deletion/insertions (Fig. 1A). Nucleotide substitutions are randomly distributed throughout sequences within a subfamily, whereas insert/deletion events are characteristic of individual subfamilies. Further, identical mutations were shared by nlRGAs from different subfamilies (Fig. 1A; arrows).

Multiple sequence alignment of the NBS domains of the sugar beet nlRGA subfamilies. For each subfamily, two sequences were chosen randomly as representatives, and the 500-bp consensus sequences flanking the P-loop and GLPL were selected. A Conserved motifs of the NBS domains are underlined. Residues that are identical among subfamilies are shaded, and nucleotide substitutions within the subfamily are boxed. Arrows indicate mutations shared by nlRGAs within or among subfamilies. Nucleotide insertions and deletions are represented by dashes. B Cladogram showing homology within and among subfamilies. Sequence lengths are shown below.
RACE amplifications were performed to isolate full-length cDNAs. From the 12 nlRGA cDNAs, four putative full-length sequences were isolated for cZR-l, -3, -7, and -9, referred to as cZR-1(f), cZR-3(f), cZR-7(f), and cZR-9(f). No RACE products were obtained from the remaining eight RGAs. A homology search revealed that the predicted products of the four full-length sugar beet nlRGAs share strong similarities with several NBS-LRR resistance genes from different species (e.g., Mi [6.5e −75, root-knot nematode resistance protein; Milligan et al. 1998], Gpa2 [5.1e −31, cyst nematode resistance protein; van der Vossen et al. 2000], Rx [1.4e −18, viral resistance protein; Bendahmane et al. 1999], I2C1 [9.8e −38, vascular wilt disease resistance protein; Ori et al. 1997], Prf and RPM1 (1.0e −25 and 1.9e −42, P. syringae resistance proteins [Grant et al. 1995; Salmeron et al. 1996]), suggesting possible roles in disease resistance responses. Because the four full-length RGAs fall into the three nlRGA groups, they were chosen as representatives for each nlRGA type in further analyses (Table 2).
Sequence Alignment Analysis
The amino acid sequences spanning the NBS domains of sugar beet nlRGAs cZR-l(f), cZR-3(f), cZR-7(f), and cZR-9(f) were aligned with 20 known NBS-LRR resistance proteins. All of the crucial motifs of the NBS domain (P-loop/kinase-1a, kinase-2, and kinase-3a, and GLPL) are highly conserved among the sugar beet nlRGAs and the known NBS-LRR resistance proteins (Fig. 2). The TIR- and non-TIR-type resistance proteins were distinguished by consensus amino acids in or around the NBS domain (Fig. 2). For example, a valine (V) and serine (S) at positions 45 and 46 are characteristic of the non-TIR-type resistance proteins, whereas a tryptophan (W) or aspartic acid (D) at position 121 is characteristic of the non-TIR-or TIR-type proteins, respectively. Differences were also detected by visual scanning of the sequence alignments at positions 320 to 327 and 373 to 376 (Fig. 2). Based on these differences, the known TIR-type resistance proteins, RPP1, RPP5, M, L6, and N, cluster into one group, called Group I by Pan et al. (2000a), whereas the non-TIR-type resistance proteins selected in this study, together with the four sugar beet nlRGAs, form a second group (Group II; Fig. 2). The remaining 28 sugar beet nlRGAs all fall into the non-TIR resistance gene family as well (data not shown).
TIR and non-TIR disease resistance proteins contain characteristic Toll-like or coiled-coil N-terminal domains. Therefore, an additional amino acid sequence alignment analysis was carried out using only the N-terminal domain. Figure 3 shows that a coiled-coil structure was predicted from the products of the four sugar beet nlRGAs with as high of a probability as that found in the known non-TIR resistance proteins. In contrast, a Toll-like consensus was found in the Group I gene products but not in the four sugar beet nlRGAs or the Group II genes (data not shown). This is consistent with the sequence alignment analysis (Fig. 2) and confirms that the four representative sugar beet nlRGAs belong to the non-TIR-type resistance gene family.

Multiple sequence alignment analysis of predicted coiled-coil domains of four representative sugar beet nlRGA proteins in comparison with those of 12 known disease resistance proteins. Probabilities of the four sugar beet nlRGAs forming a coiled-coil structure in their N-terminal domains, using the COILS (Lupas, 1996) and PEPCOIL prediction programs (Institute of Pasteur), are: cZR-1(f), 2 regions with p(7) = 1.0; cZR-3(f), p(21) = 0.6, but for five regions, p(7) = 1.0; cZR-7(f), p(28) = 1.0; cZR-9(f), p(21) = 0.7. The hepated repeat, where the seven positions are labeled from a to g, is displayed below the sequences in the coiled-coil domains. Positions a and d are boxed.
Phylogenetic Analysis
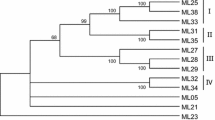
Twenty known disease resistance genes and the four representative sugar beet nlRGAs were pooled for a phylogenetic analysis. A phylogenetic tree was created on the basis of a 400-amino acid sequence spanning the NBS domains using the neighbor-joining method (Saitou and Nei 1987). In accordance with previous reports (Pan et al. 2000a; Cannon et al. 2002), two major branches were found in the phylogenetic tree (Fig. 4): the TIR- and non-TIR-type disease resistance proteins. The four sugar beet nlRGAs fall in the non-TIR branch but are distributed into two subbranches that probably represent divergent recent ancestral genes. Thus, cZR-1(f) and cZR-9(f) appear to have arisen from an ancestor common to Prf, Rx, RPP8, and Mi, whereas cZR-3(f) and cZR-7(f) originated from the ancestral gene from which I2C1, Xa1, and Cre3 arose (Fig. 4).

Phylogenetic tree of the sugar beet nlRGAs and other disease resistance genes based on the 400-amino acid sequence of the NBS domain. Sequences were aligned using ClustalW, and the tree wasgenerated using the neighbor-joining method. Numbers on the branches indicate percentage bootstrap replications (1000). Arrows indicate the location of sugar beet nlRGAs in the tree. Groups I and IIcorrespond to TIR and non-TIR disease resistance gene subfamilies, respectively (Pan et al. 2000b).
Analysis of Sugar Beet EST Databases
To address whether TIR-type nlRGAs can be found in current sugar beet EST databases, we extended our analyses to search more than 16,000 publicly available beet ESTs (released on 15 December 2002). The majority of these ESTs were isolated from a normalized cDNA library made from sugar beet leaf, developing root, storage root, and inflorescence (Herwig et al. 2002). Two NBS consensus sequences, TIR and non-TIR NBS-HMM, were generated and used as queries. Nine significant nonredundant hits with E values less than e −08 were returned (BI543249.1, BQ060540.1, BQ060608, BQ585814, BQ591373, BQ584922, BQ593129, BQ584971, and BQ585687). All nine, however, were homologues retrieved with the non-TIR NBS-HMM. No representative TIR NBS-HMM sequences were detected (Table 3). To prove the specificity of the queries used, we tested them against EST databases of rice, Arabidopsis, and cedar (Cryptomeria japonica). Significant non-TIR-type hits were obtained from all three databases, whereas TIR-type homologues were detected only in the dicot plants, A. thaliana and C. japonica, and not in the monocot rice (Table 3). These results were consistent with those noted by Cannon et al. (2002). Using the same queries, we found both TIR- and non-TIR-type RGAs in the cedar EST database, which contains only 2438 sequences, roughly one-seventh of the beet EST database used in this study. Thus, the number of EST sequences available in the beet databases is probably not a limiting factor in this study.
To verify our search profiles, a TIR consensus sequence was generated on the basis of the N-terminal sequences of the disease resistance proteins from Group I (Fig. 2) and used to query the same EST databases used in the NBS-HMM searches. Table 4 shows that significant hits were found in the two dicot plants, with more than 100 from Arabidopsis and 2 from cedar, respectively. In comparison, only 30 hits from Arabidopsis and 2 from cedar were found in common in the NBS-HMM and TIR consensus queries. This variation probably reflects the dynamic evolution of NBS-LRR disease resistance proteins. This is in accordance with the results reported by Jebanathirajah et al. (2002) that at least four proteins in the Arabidopsis genome contain only TIR domains without any other identifiable domains.
The search using the TIR consensus sequence query revealed one additional TIR type homologue sequence from sugar beet and four from rice EST databases (beet: BQ585814 with E-value e −24; rice: AP003866.2, AAAA01012636.1, AAAA01000037.1, and CL012502.49 with E-value 7e −04). However, upon further inspection of the sequence, we were unable to find the conserved NBS domain following the TIR regions. Therefore, we concluded that these sequences probably represent another type of TIR-containing sequence (Jebanathirajah et al. 2002) or a vestige of the TIR-type NBS-LRR resistance sequences that were lost throughout evolution. The latter possibility has also been discussed as possible for the rice genome (Bai et al. 2002).
We extended the database search into two other Chenopodiaceae species, Spinacia oleracea and Suaeda maritima, for which sequence data are available in GenBank (400 nucleotide and 1291 protein entries are available for Spinacia, and 1008 nucleotide and 1000 EST entries are available for Suaeda). Search results were negative for both NBS-HMM and TIR consensus sequences (data not shown).
Compositional Gradients of GC Content and Codon and Amino Acid Usage
The similar lack of TIR-type disease resistance genes in sugar beet and in cereals led us to speculate on a possible synteny among these genomes. To this end, we compared the composition of the genomes, including GC content, codon and amino acid usage, of sugar beet genes with that of monocots and dicots. One recently described property of genes in the Gramineae is characteristic of gradients in GC content, codon usage, and amino acid usage along the direction of transcription, beginning at the junction of the 5′-UTR and the coding region (Wong et al. 2002; Yu et al. 2002).
The compositional gradients of GC content, codon, and amino acid usage were compared using the coding sequences (CDS) downloaded from the Codon Usage Database (http://www.kazusa.or.jp/codon) according to the method described by Yu et al. (2002). Figure 5 shows that a gradient of GC content along the codon region was observed in the cereal genes but not in the sugar beet or dicot genes. The pattern of GC content in the sugar beet genes is clearly divergent from that in the cereals but analogous to that in other dicot species (Fig. 5A). Similarly, a gradient of GC content appeared along the coding region of the resistance gene Xa1, cloned from the monocot rice, but not in the dicot sugar beet RGAs, cZR-7, or the Arabidopsis RPM1 (Fig. 5B). Comparison of GC contents at the three codon positions (GC1/GC2/GC3) among different species revealed that sugar beet shares the same GC content order with other dicots. The relative magnitudes of GC content were GC1 > GC2 > GC3, which differs from those of the cereal genes (GC3 > GC1 ≈ GC2) (Fig. 5C). These results are consistent with those reported by Wong et al. (2002). We also attempted to compare the frequencies of serines and alanines at the 5′ ends of gene products (including methionine starts and more than 60 amino acids) from sugar beet, rice, and Arabidopsis. Serine and alanine enrichments at the 5′ ends of dicot and Gramineae genes, as reported by Yu et al. (2002), however, were not found in this study, and only a small serine peak appeared at the 5′ ends of the sugar beet genes (Fig. 5D).

Analysis of compositional gradients of GC content, codon usage, and amino acid usage of sugar beet genes in comparison with cereal and dicot plant genes. A Comparative analysis of GC content as a function of cDNA position, relative to the start of the coding region. The data shown are an average of more than 70 coding sequences from sugar and from each of five cereals as well as of seven dicot plants. B Comparative analysis of GC content with the predicted coding sequences of cZR-7 from sugar beet, Xa1 from rice,and RPM1 from Arabidopsis. A 200-bp sliding window was used to filter out the fluctuations in the sequence; C Analysis of GC content at three coding positions of genes from sugar beet in comparison with those of monocot cereal and dicot plant genes. Shown here are an average of more than 70 coding sequences from sugar beet and from each of five cereals as well as of seven dicot plants. D Frequency of occurrence for sugar beet alanine and serine as a function of cDNA position, relative to the start of the coding region, and averaged overall protein with a 17-amino acid (AA) sliding window. When all 20 AA occur with equal probability, the normalized frequencies are 1. A total of 25 protein sequences was used.
In addition, correlation analysis of codon usage on the basis of the CDS from different species revealed a significant coefficient of codon usage among sugar beet and dicot genes but not among sugar beet and cereal genes (p < 0.05) (Table 5). To determine possible differences in codon usage within resistance genes or RGAs, the four full-length sugar beet nlRGAs, four known resistance genes from rice, and five from Arabidopsis were compiled into a table for comparison. In this context, sugar beet is again more similar to Arabidopsis than to rice and thus falls into the same category as dicot plants.
Discussion
Here we report the isolation and phylogenetic analyses of sugar beet NBS-LRR resistance gene analogues (nlRGAs), demonstrating the potential of PCR-based approaches for cloning resistance gene homologues from sugar beet. We combined a PCR-based approach with a cDNA library screening and identified 27 genomic clones and 12 expressed sequences of NBS-LRR RGAs, including four full-length RGA sequences. These RGAs, along with available sugar beet EST databases, enabled us to gain the first insights into the evolution of NBS-LRR-type disease resistance genes in sugar beet.
Various genetic mechanisms have been discussed to account for the evolution of R-genes. These mechanisms include a slowly evolving divergence hypothesis (Michelmore and Meyers 1998; Stahl et al. 1999) and a rapidly evolving process (Leister et al. 1998; McDowell et al. 1998). Abundant data also indicate that R-genes and homologous sequences are clustered in plant genomes. It is believed that clustered RGAs are more likely than isolated genes to be strongly shaped by frequent gene duplication, loss, conversion, and ectopic recombination, thus facilitating the generation of diversity and specificity in new R-genes (Ronald et al. 1998). The sugar beet nlRGAs isolated in this study showed considerable sequence variation. However, they clustered into at least three subfamilies. Sequences from within one class share greater than 90% homology; in contrast, those from different subfamilies share less than 65% identity. Most likely, the former sequences represent gene families from a recent common origin, whereas the latter represent diverged unique origins. Comparative sequence analyses within each of the three subfamilies suggest that point mutations (substitutions, insertions/deletions) may be the primary source of divergence of the sugar beet nlRGAs. Thus, in sugar beet a gradual accumulation of mutations rather than a rapidly evolving process apparently accounts for the divergence of resistance mechanisms against dynamically developing pathogen populations, as suggested by Michelmore and Meyers (1998) and Stahl et al. (1999). Considering the high diversity of non-TIR-type RGAs in plant genomes (Bai et al. 2002 and Cannon et al. 2002), additional non-TIR-type RGAs from sugar beet should be tested to confirm this hypothesis.
The striking sequence similarities among sugar beet nlRGAs and R-genes from different angiosperm species suggest the existence of an ancient common ancestor of disease resistance genes. The fact that the sugar beet nlRGAs that were isolated fell into two diverged subbranches in the phylogenetic tree imply functional divergence of these sequences in resistance responses, e.g., either in recognition of pathogen elicitors or in activating signal transduction pathways (Aarts et al. 1998b). Taking into account that the four full-length nlRGAs were cloned from a nematode-resistant sugar beet line and all show a high homology to known resistance proteins against nematode and virus, respectively, thus functional analyses of the four full-length nlRGAs, with respect to their roles in initiating or amplifying various disease resistance responses to nematode or rhizomania, e.g., may contribute significantly to the elucidation of the possible mechanism.
An important finding of this study is that all nlRGAs isolated fall into the non-TIR NBS-LRR resistance gene subfamily. The predicted protein products of nlRGAs contain not only the consensus amino acid usage for non-TIR-type NBS-domain, but also the potential to form coiled-coil structures at their N-termini, a characteristic of the non-TIR-type resistance gene subfamily. In addition to screening of a sugar beet cDNA library using a probe that exhibits high homology at the nucleotide level to both TIR and non-TIR types of resistance genes, homology searches using either TIR NBS-HMM or TIR consensus sequences as queries did not return any representatives of the TIR NBS-LRR resistance gene subfamily from the beet EST databases, though they were readily detected in other dicot EST databases that served as controls, e.g., the small cedar EST database, which contains only 2438 sequences. Also, the four TIR type-specific primers failed to amplify any TIR-type nlRGAs from the sugar beet genome. These data suggest an absence, rather than low expression levels, of TIR-type NBS-LRR resistance gene homologues in the sugar beet genome. A similar result was recently reported by Hunger et al. (2003).
Although the TIR and non-TIR/coiled-coil termini probably function analogously in recruiting downstream components through protein–protein interactions and activation of pathogen-dependent resistance responses, it was suggested that the common ancestor of plant NBS-LRR resistance genes, which predates the divergence of angiosperms and gymnosperms, included a TIR domain (Bold 1977; Meyers et al. 1999; Pan et al. 2000a). The TIR domain is highly conserved across species, and a moderate sequence similarity is also present in domains of plant NBS-LRR-type disease resistance genes, the Drosophila Toll/Interleukin-1 receptor (IL-1R), and the Apaf-1 and Ced-4 genes. The latter genes are implicated in regulating the animal immune response and apoptosis, suggesting that regulatory mechanisms of the cell death programs are conserved among plants, mammals, and other animals. Nevertheless, the number of genes encoding proteins that contain TIR domains in Arabidopsis (about 135) is over 10 times that found in human, Drosophila, or C. elegans genomes (Jebanathirajah et al. 2002). This difference indicates that expansion, as well as clustering, of these sequences in plant genomes was required throughout evolution to enhance defense responses to different pathogens.
One reasonable explanation for the absence of TIR NBS-LRR disease resistance RGAs in the sugar beet genome is the loss of this type of sequence over time. This loss also presumably occurred in cereal (Meyers et al. 1999; Pan et al. 2000a; Cannon et al. 2002), although no mechanism has been described to explain the elimination of a dispersed multigene family from a plant genome. The modern diversity of NBS-LRR resistance genes is thought to have arisen from a few ancestral loci. The expansion of a limited number of ancestral genes, which differed among plant lineages, may have resulted in either degeneration or complete loss of certain loci (Pan et al. 2000a; Noir et al. 2001). Indeed, the loci encoding TIR NBS-LRR resistance genes have been lost, whereas the non-TIR-type resistance sequences have expanded and diversified throughout evolution. This scenario fits well with the uneven distribution of NBS-LRR sequences in plant lineages and large copy numbers of genes in modern species (Pan et al. 2000a; Fluhr 2001). A complete loss of TIR sequences in cereals and in coffee trees (Coffea L.) has been explained as dramatic examples of this scenario (Pan et al. 2000a; Noir et al. 2001). Eventually, the occasional and random mutational events of the ancestral loci, as found in sugar beet RGAs in this study, may have contributed to the disappearance of TIR NBS-LRR resistance genes in plant genomes.
This finding leads to the assumption that TIR NBS-LRR genes could also have been lost from other dicot species. Figure 6 illustrates a modified version of an evolution model of plant disease resistance genes presented by Pan et al. (2000a). At least two stages are involved in the evolution of NBS-LRR resistance genes in the plant kingdom: the presence of a few NBS-LRRs with broad spectrum specificity (Stage I) and the presence of resistance genes shaped by divergent gene duplication followed by gene diversification after the monocot/dicot separation (Stage II). The loss of TIR type RGAs in the sugar beet genome implies that the loss of TIR-type resistance sequences is not restricted to cereals or monocots in general and that the loss of TIR NBS-LRR genes in cereal species should not be generalized to all monocots. It was suggested that the loss of resistance gene clades was a result of either the loss of monocot-specific RGAs from certain dicot families or the creation of novel sequences in monocots, thus providing instances of “birth” and “death” of ancient lineages of RGAs within particular plant families (Cannon et al. 2002). The fact that we did not find any synteny among sugar beet and cereals at the nucleotide or amino acid levels supports the idea that sequences were indeed lost from those ancestral loci in plant genomes, regardless of the type of species. It is tempting to speculate that these changes reflect differences in downstream defence signaling pathways (Bai et al. 2002).

A modified version of a model by Pan et al. (2000a) of the evolution of NBS-LRR-type disease resistance genes. At least two stages are involved in the evolution of NBS-LRR-type resistance genes. During Stage I, a few NBS-LRRs with broad spectrum specificity were present. Stage II involved divergent gene duplication followed by gene diversification after the monocot/dicot separation. During Stage II, TIR NBS-LRR genes were lost from cereal and sugar beet genomes.
The loss of TIR-type NBS-LRR sequences in sugar beet has practical implications for the potential use of resistance genes in sugar beet and for the understanding of the evolution of RGAs in general, as the TIR and the non-TIR NBS-LRR genes trigger disease resistance responses via different signal transduction pathways. In Arabidopsis, the TIR-type resistance genes require a functional EDS1 allele to activate hypersensitive cell death disease responses, whereas the non-TIR-type resistance genes require a functional NDR1 allele to activate disease resistance (Parker et al. 1996; Century et al. 1997; Aarts et al. 1998b). Therefore, further genetic and biochemical analyses are required to determine whether a mechanism analogous to that used by TIR type resistance genes exists in sugar beet or Chenopodiaceae. Also, the time of the presumed loss of the TIR type resistance genes in sugar beet remains to be determined; alternatively, other hypotheses may be tested. It will be particularly interesting to determine the genomic organization of nlRGA-clusters and their expression patterns in relation to disease resistance responses, e.g., against rhizomania and nematodes.
References
MG Aarts B Hekkert EB Holub JL Beynon WJ Stiekema A Pereira (1998a) ArticleTitleIdentification of R-gene homologous DNA fragments genetically linked to disease resistance loci in Arabidopsis thaliana. Mol Plant Microbe Interact 11 251–258 Occurrence Handle1:CAS:528:DyaK1cXitVGqsLc%3D
N Aarts M Metz E Holub BJ Staskawicz MJ Daniels JE Parker (1998b) ArticleTitleDifferent requirements for EDS1 and NDR1 by disease resistance genes define at least two R gene-mediated signalling pathways in Arabidopsis. Proc Natl Acad Sci USA 95 10306–10311 Occurrence Handle1:CAS:528:DyaK1cXlsFGgsb4%3D
SF Altschul TL Madden AA Schaffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25 3389–3402 Occurrence Handle9254694
L Aravind VM Dixit EV Koonin (1999) ArticleTitleThe domains of death: Evolution of the apoptosis machinery. Trends Biochem Sci 24 47–53 Occurrence Handle10.1016/S0968-0004(98)01341-3 Occurrence Handle1:CAS:528:DyaK1MXltFChu7g%3D Occurrence Handle10098397
K Arumuganathan ED Earle (1991) ArticleTitleNuclear DNA content of some important plant species. Plant Mol Biol Rep 9 208–218 Occurrence Handle1:CAS:528:DyaK38XhslSjurY%3D
JF Bai LA Pennill J Ning SW Lee R Jegadeesan CR Webb BY Zhao Q Sun JC Nelson JE Leach SH Hulbert (2002) ArticleTitleDiversity in nucleotide binding site-Leucine-rich repeat genes in cereals. Genome Res 12 1871–1884 Occurrence Handle10.1101/gr.454902 Occurrence Handle1:CAS:528:DC%2BD38XpsVOqtbc%3D Occurrence Handle12466291
B Baker P Zambryski B Staskawicz SP Dinesh-Kumar (1997) ArticleTitleSignalling in plant-microbe interactions. Science 276 726–733 Occurrence Handle1:CAS:528:DyaK2sXivFOltLg%3D Occurrence Handle9115193
A Bendahmane K Kanyuka DC Baulcombe (1999) ArticleTitleThe Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11 781–792 Occurrence Handle1:CAS:528:DyaK1MXjvVWrtLc%3D Occurrence Handle10330465
HC Bold (1977) The plant kingdom. Prentice-Hall Englewood Cliffs, NJ
SB Cannon H Zhu AM Baumgarten R Spangler G May DR Cook ND Young (2002) ArticleTitleDiversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS-LRR resistance gene subfamilies. J Mol Evol 54 548–562 Occurrence Handle10.1007/s00239-001-0057-2 Occurrence Handle1:CAS:528:DC%2BD38Xjt1Ggu70%3D Occurrence Handle11956693
KS Century AD Shapiro PP Repetti D Dahlbeck E Holub BJ Staskawicz (1997) ArticleTitle NDR1, a pathogen-induced component required for Arabidopsis disease resistance. Science 278 1963–1965 Occurrence Handle1:CAS:528:DyaK2sXotVClt78%3D Occurrence Handle9395402
JL Dangl JD Jones (2001) ArticleTitlePlant pathogens and integrated defence responses to infection. Nature 411 826–833 Occurrence Handle1:CAS:528:DC%2BD3MXksF2gu74%3D Occurrence Handle11459065
JG Ellis GJ Lawrence JE Luck PN Dodds (1999) ArticleTitleIdentification of regions in alleles of the flax rust resistance gene L that determine differences in gene-for-gene specificity. Plant Cell 11 495–506 Occurrence Handle1:CAS:528:DyaK1MXitlehsLo%3D Occurrence Handle10072407
R Fluhr (2001) ArticleTitleSentinels of disease. Plant resistance genes. Plant Physiol 127 1367–1374 Occurrence Handle1:CAS:528:DC%2BD38XjtVWiug%3D%3D Occurrence Handle11743075
MR Grant L Godiard E Straube T Ashfield J Lewald A Sattler RW Innes JL Dangl (1995) ArticleTitleStructure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 269 843–846 Occurrence Handle1:CAS:528:DyaK2MXnsVagsrg%3D Occurrence Handle7638602
KE Hammond-Kosack JD Jones (1997) ArticleTitlePlant disease resistance genes. Annu Rev Plant Physiol Plant Mol Biol 48 575–607 Occurrence Handle1:CAS:528:DyaK2sXjs1elsr0%3D
R Herwig B Schulz B Weisshaar S Hennig M Steinfath M Drungowski D Stahl W Wruck A Menze J O’Brien H Lehrach U Radelof (2002) ArticleTitleConstruction of a “unigene” cDNA clone set by oligonucleotide fingerprinting allows access to 25,000 potential sugar beet genes. Plant J 32 845–857 Occurrence Handle10.1046/j.1365-313X.2002.01457.x Occurrence Handle12472698
T Horng GM Barton R Medzhitov (2001) ArticleTitleTIRAP: An adapter molecule in the Toll signalling pathway. Nat Immunol 2 835–841 Occurrence Handle10.1038/ni0901-835 Occurrence Handle1:CAS:528:DC%2BD3MXmsFehtrc%3D Occurrence Handle11526399
S Hunger G Di Gaspero S Mohring D Bellin R Schafer-Pregl DC Borchardt CE Durel M Werber B Weisshaar F Salamini K Schneider (2003) ArticleTitleIsolation and linkage analysis of expressed disease-resistance gene analogues of sugar beet (Beta vulgaris L.). Genome 46 70–82 Occurrence Handle10.1139/g02-106 Occurrence Handle12669798
J Jebanathirajah S Peri A Pandey (2002) ArticleTitleToll and interleukin-1 receptor (TIR) domain-containing proteins in plants: A genomic perspective. Trends Plant Sci 7 388–391 Occurrence Handle10.1016/S1360-1385(02)02309-9 Occurrence Handle1:CAS:528:DC%2BD38XntVWnu7s%3D Occurrence Handle12234729
DA Jones JDG Jones (1997) ArticleTitleThe role of leucine-rich repeat proteins in plant defences. Adv Bot Res 24 90–167
D Leister J Kurth DA Laurie M Yano T Sasaki K Devos A Graner P Schulze-Lefert (1998) ArticleTitleRapid reorganization of resistance gene homologues in cereal genomes. Proc Natl Acad Sci USA 95 370–375 Occurrence Handle1:CAS:528:DyaK1cXjtl2gsg%3D%3D Occurrence Handle9419382
A Lupas (1996) ArticleTitleCoiled coils: New structures and new functions. Trends Biochem Sci 21 375–382 Occurrence Handle1:CAS:528:DyaK28XmsleitLs%3D Occurrence Handle8918191
JM McDowell M Dhandaydham TA Long MG Aarts S Goff EB Holub JL Dangl (1998) ArticleTitleIntragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of Arabidopsis. Plant Cell 10 1861–1874 Occurrence Handle9811794
BC Meyers AW Dickerman RW Michelmore S Sivaramakrishnan BW Sobral ND Young (1999) ArticleTitlePlant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20 317–332 Occurrence Handle10.1046/j.1365-313X.1999.00606.x Occurrence Handle1:CAS:528:DC%2BD3cXjsF2kug%3D%3D Occurrence Handle10571892
RW Michelmore BC Meyers (1998) ArticleTitleClusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8 1113–1130 Occurrence Handle1:CAS:528:DyaK1cXotVWhurg%3D Occurrence Handle9847076
SB Milligan J Bodeau J Yaghoobi I Kaloshian P Zabel VM Williamson (1998) ArticleTitleThe root knot nematode resistance gene Mi from tomato is a member of the leucine zipper, nucleotide binding, leucine-rich repeat family of plant genes. Plant Cell 10 1307–1319 Occurrence Handle1:CAS:528:DyaK1cXls1Sis7s%3D Occurrence Handle9707531
L Noel TL Moores EA van Der Biezen M Parniske MJ Daniels JE Parker JD Jones (1999) ArticleTitlePronounced intraspecific haplotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell 11 2099–2112 Occurrence Handle10559437
S Noir MC Combes F Anthony P Lashermes (2001) ArticleTitleOrigin, diversity and evolution of NBS-type disease-resistance gene homologues in coffee trees (Coffea L.). Mol Genet Genomics 265 654–662 Occurrence Handle10.1007/s004380100459 Occurrence Handle1:CAS:528:DC%2BD3MXlsV2gurc%3D Occurrence Handle11459185
N Ori Y Eshed I Paran G Presting D Aviv S Tanksley D Zamir R Fluhr (1997) ArticleTitleThe I2C family from the wilt disease resistance locus I2 belongs to the nucleotide binding, leucine-rich repeat superfamily of plant resistance genes. Plant Cell 9 521–532 Occurrence Handle1:CAS:528:DyaK2sXivFCltbw%3D Occurrence Handle9144960
RD Page (1996) ArticleTitleTree View: An application to display phylogenetic trees on personal computers. Comput Appl Biosci 12 357–358 Occurrence Handle1:STN:280:ByiD2MfgtlA%3D Occurrence Handle8902363
Q Pan J Wendel R Fluhr (2000a) ArticleTitleDivergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50 203–213 Occurrence Handle1:CAS:528:DC%2BD3cXivVKgt7k%3D
Q Pan YS Liu O Budai-Hadrian M Sela L Carmel-Goren D Zamir R Fluhr (2000b) ArticleTitleComparative genetics of nucleotide binding site-leucine rich repeat resistance gene homologues in the genomes of two dicotyledons: tomato and Arabidopsis. Genetics 155 309–322 Occurrence Handle1:CAS:528:DC%2BD3cXjslKhtL0%3D
J Park K Karplus C Barrett R Hughey D Haussler T Hubbard C Chothia (1998) ArticleTitleSequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol 284 1201–1210 Occurrence Handle10.1006/jmbi.1998.2221 Occurrence Handle1:CAS:528:DyaK1MXivFyrsg%3D%3D Occurrence Handle9837738
JE Parker EB Holub LN Frost A Falk ND Gunn MJ Daniels (1996) ArticleTitleCharacterization of eds1, a mutation in Arabidopsis suppressing resistance to Peronospora parasitica specified by several different RPP genes. Plant Cell 8 2033–2046
MI Rivkin CE Vallejos PE McClean (1999) ArticleTitleDisease-resistance related sequences in common bean. Genome 42 41–47 Occurrence Handle1:CAS:528:DyaK1MXhsVyqsbs%3D Occurrence Handle10208000
SO Rogers AJ Bendich (1985) ArticleTitleExtraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol Biol 5 69–76 Occurrence Handle1:CAS:528:DyaL2MXlvFalsrk%3D
PC Ronald (1998) ArticleTitleResistance gene evolution. Curr Opin Plant Biol 1 294–298
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic tree. Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
JM Salmeron GE Oldroyd CM Rommens SR Scofield HS Kim DT Lavelle D Dahlbeck BJ Staskawicz (1996) ArticleTitleTomato Prf is a member of the leucine-rich repeat class of plant disease resistance genes and lies embedded within the Pto kinase gene cluster. Cell 86 123–133 Occurrence Handle1:CAS:528:DyaK28XktlGnsbs%3D Occurrence Handle8689679
F Sanger S Nicklen AR Coulson (1977) ArticleTitleDNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74 5463–5467 Occurrence Handle271968
KA Shen BC Meyers MN Islam-Faridi DB Chin DM Stelly RW Michelmore (1998) ArticleTitleResistance gene candidates identified by PCR with degenerate oligonucleotide primers map to clusters of resistance genes in lettuce. Mol Plant Microbe Interact 11 815–823 Occurrence Handle1:CAS:528:DyaK1cXkslWht7k%3D Occurrence Handle9675895
E Speulman D Bouchez EB Holub JL Beynon (1998) ArticleTitleDisease resistance gene homologs correlate with disease resistance loci of Arabidopsis thaliana. Plant J 14 467–474 Occurrence Handle1:CAS:528:DyaK1cXkt1Khtbc%3D Occurrence Handle9670562
EA Stahl G Dwyer R Mauricio M Kreitman J Bergelson (1999) ArticleTitleDynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400 667–671 Occurrence Handle1:CAS:528:DyaK1MXlsVCktb8%3D Occurrence Handle10458161
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
GM Timmerman-Vaughan TJ Frew N Weeden (2000) ArticleTitleCharacterization and linkage mapping of R-gene analogous DNA sequences in pea (Pisum sativum L.). Theor Appl Genet 101 241–247 Occurrence Handle1:CAS:528:DC%2BD3cXlvVyksbg%3D
TW Traut (1994) ArticleTitleThe functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur J Biochem 222 9–19 Occurrence Handle1:CAS:528:DyaK2cXktVajtrY%3D Occurrence Handle8200357
EA van der Biezen JD Jones (1998) ArticleTitleThe NB-ARC domain: A novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr Biol 8 226–227
der van EA Biezen J Sun MJ Coleman MJ Bibb JD Jones (2000) ArticleTitle Arabidopsis RelA/SpoT homologs implicate (p)ppGpp in plant signalling. Proc Natl Acad Sci USA 97 3747–3752 Occurrence Handle10.1073/pnas.060392397 Occurrence Handle10725385
EA van der Vossen JN van der Voort K Kanyuka A Bendahmane H Sandbrink DC Baulcombe J Bakker WJ Stiekema RM Klein-Lankhorst (2000) ArticleTitleHomologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: A virus and a nematode. Plant J 23 567–576 Occurrence Handle10972883
ZX Wang M Yano U Yamanouchi M Iwamoto L Monna H Hayasaka Y Katayose T Sasaki (1999) ArticleTitleThe Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J 19 55–64 Occurrence Handle10.1046/j.1365-313X.1999.00498.x Occurrence Handle10417726
GK Wong J Wang L Tao J Tan J Zhang DA Passey J Yu (2002) ArticleTitleCompositional gradients in Gramineae genes. Genome Res 12 851–856 Occurrence Handle10.1101/gr.189102 Occurrence Handle1:CAS:528:DC%2BD38Xks12hsr8%3D Occurrence Handle12045139
J Yu S Hu J Wang et al. (2002) ArticleTitleA draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296 79–92 Occurrence Handle1:CAS:528:DC%2BD38XivVSqtr8%3D Occurrence Handle11935017
Acknowledgements
This work was financially supported by the EU (FAIR6-CT08-4235) and the DFG (CA220/2-2). Dr. Fan Longjian thanks the Christian-Albrechts-University, Kiel, Germany for travel grants. We also thank Ms. C. Thiele for technical assistance and Dr. Nikki LeBrasseur for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tian, Y., Fan, L., Thurau, T. et al. The Absence of TIR-Type Resistance Gene Analogues in the Sugar Beet (Beta vulgaris L.) Genome . J Mol Evol 58, 40–53 (2004). https://doi.org/10.1007/s00239-003-2524-4
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2524-4