
Abstract
Various retroviruses have been shown to encode dUTPase. The overall phylogeny of dUTPase is unclear, though. The human genome contains a significant amount of human endogenous retroviruses (HERV) representing fossilized sequences of ancient exogenous retroviruses. A few HERV families have been reported to harbor dUTPase domains. We surveyed the various HERV families for the presence of dUTPase and found that ancestors of all HERV-K families but one encoded dUTPase. With two exceptions phylogenetic analysis shows a monophyletic origin of dUTPase for the different HERV-K dUTPases. Sequences of consensus dUTPase domains suggest that the various exogenous ancestors of HERV-K once encoded active enzymes. Our analysis provides informations on dUTPase phylogeny and further shows that endogenous retroviruses provide important informations regarding retrovirus evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human genome comprises approximately 8% of sequences of retroviral origin that stem from infections of germ cell genomes by distinct exogenous retroviruses many million years ago. The proviral sequences were subsequently fixed in the evolutionary lineage leading to humans. Many of the so-called human endogenous retroviruses (HERVs) were fixed in the germ line of Old World monkeys immediately after the evolutionary separation from New World monkeys, about 35 million years ago. About 100 different HERV families have been defined. Some families increased their proviral copy number up to several thousand copies by what is thought to be intracellular, retrovirus-like retrotransposition, that is, the formation of new proviruses by reverse transcription of a retroviral RNA in a retrovirus-like fashion (Coffin et al. 1997; International Human Genome Sequencing Consortium 2001; Jurka 2000; Lower et al. 1996).
Unfortunately, there is, as of yet, no established nomenclature for HERVs. The various HERV families have been named according to the tRNA that was once used during reverse transcription. Several HERV families utilized a lysin-tRNA and are therefore named HERV-K. Medstrand and Blomberg (1993) showed a phylogenetic relationship between the various HERV-K families based on sequence comparisons of a conserved reverse transcriptase region. The different families were named human MMTV-like (HML-1 to HML-6) because of sequence similarities to the mouse mammary tumor virus. The same group subsequently extended the number of distinct HERV-K families to nine (Andersson et al. 1999). The database for repetitive sequences, Repbase, defines 10 HERV-K families. The HERV-K family definitions are almost consistent, yet a different nomenclature is employed (Jurka 2000). The various HERV-K families integrated into the germ line of Old World primates about 35 million years ago, immediately after their evolutionary separation from New World primates.
Besides basic retroviral enzymatic functions, such as protease, reverse transcriptase, RNaseH, and endonuclease/integrase, several retroviruses encode a protein domain for dUTPase (EC 3.6.1.23) that catalyzes the hydrolysis of dUTP to dUMP and pyrophosphate. dUMP serves as a precursor for TTP synthesis, while, equally important, the cellular dUTP concentration is strongly reduced to prevent incorporation of excess and mutagenic dUTP into newly synthesized DNA. That dUTPase is an important enzyme is evident from its ubiquitous presence in eukaryotes, eubacteria, and archaea. dUTPase activity in various viruses has been reported to be beneficial during replication of viral genomes in both dividing and nondividing host cells (Oliveros et al. 1999; Turelli et al. 1996). Several retroviruses related to MMTV and nonprimate lentiviruses encode dUTPase. Horizontal transfer of dUTPase between those two groups has been suggested recently, as opposed to independent acquisition from a cellular source. However, the overall phylogeny of dUTPase is still subject to speculation (Baldo and McClure 1999; Vassylyev and Morikawa 1996). dUTPase motifs were also identified in a few human endogenous retroviruses, e.g., the foamy virus-related HERV-L family for which dUTPase is located downstream from the endonuclease/integrase domain (Cordonnier et al. 1995). Proviruses belonging to the HERV-K(HML-2) family also contain dUTPase motifs located N-terminal to the protease domain. For that family the conserved motif 5 displays mutations resulting in loss of enzymatic activity that could be restored when essential amino acid positions were corrected (Harris et al. 1997). Cryptic dUTPase motifs have been reported for proviral fragments of the HERV-K(HML-6) and HERV-K(HML-5) families (Tristem 2000; Yin et al. 1999). We recently showed the presence of a dUTPase domain in a HERV-K(HML-3) consensus sequence (Mayer and Meese 2002).
In the present study we asked whether all HERV-K families once encoded dUTPase and whether other HERV families also harbored dUTPase motifs. All HERV-K families but one displayed dUTPase motifs. Our study further demonstrates that HERVs as “sequence fossils” of former exogenous retroviruses, having targeted the primate lineage about 35 million years ago, can still provide sufficient information to deduce intact retroviral sequences and thus retroviral motifs, genes, and proteins from putative former exogenous retroviruses.
Materials and Methods
Collecting HERV-K Proviruses from the Human Genome Sequence
We used the previously generated HERV-K consensus sequences from Repbase (Jurka 2000) to perform BLAT-Searches on the respective latest freeze versions of the Human Genome Browser (http://genome.ucsc.edu ) (Kent et al. 2002) using standard parameters. We downloaded each provirus plus flanking sequence and compared its overall structure to that of the respective Repbase HERV-K consensus sequence by dot matrix comparisons.
Identifying Protease Corresponding Regions
For each Repbase HERV-K sequence we delineated the protease gene region from sequence similarities to known retroviral proteases as revealed by BlastX at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov ). Using the above-mentioned dot matrix comparisons we projected the protease region plus about 200 bp on each side onto each provirus sequence. The respective protease region was extracted from each provirus sequence provided the region was present. Within the extracted portions we identified other repeats, such as Alu or L1 elements, by RepeatMasker (A.F.A. Smit and P. Green, unpublished data; http://ftp.genome.washington.edu/cgi-bin/RepeatMasker ) and deleted those elements before multiple alignments.
Multiple Alignment and Generation of Consensus Sequences
We aligned sequences for each HERV-K family using ClustalW (Thompson et al. 1994) and standard parameters. We refined alignments by hand using the Se-A1 program (provided by Andrew Rambaut, University of Oxford, Oxford, UK). We generated consensus sequences from multiple alignments using the Boxshade server at the Institut Pasteur (http://bioweb.pasteur.fr/seqanal/interfaces/boxshade.html ). We corrected ambiguous positions in the consensus sequence by hand according to the majority rule and with regard to open reading frames.
Identification of dUTPase Motifs and Phylogenetic Analysis
We analyzed the dUTPase/protease consensus sequences for open reading frames. The longest resulting reading frame was translated into a protein sequence that was subjected to search for conserved domains employing CD-search at the NCBI (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi ). HERV families other than HERV-K were examined for the presence of dUTPase motifs by three-phase translation of consensus/reference sequences, as they were included in Repbase, and subsequent CD-search for (cryptic) dUTPase motifs within the resulting protein sequences. Regenerated dUTPase protein sequences were multiple aligned employing a previously established hidden Markov model profile for dUTPases (pfam00692) obtained from the Pfam Database (http://pfam.wustl.edu ) and using the hmmalign program provided by the Institut Pasteur (Eddy 1998; Sonnhammer et al. 1998) (http://bioweb.pasteur.fr/seqanal/interfaces/hmmalign.html ).
The HERV-K dUTPase protein sequences were phylogenetically compared to other dUTPases included in the dUTPase pfam dataset (Bateman et al. 2002) by different methods. Following the recent analysis by Baldo and McClure (1999) we calculated distances between sequences using ProtDIST and employing a Dayhoff PAM matrix. One hundred bootstraps were analyzed by the Fitch Margoliash algorithm (Fitch, with jumbling three times each) and a consensus tree was generated. Furthermore, we analyzed phylogenetic relationships employing the Parsimony (PAUP*; Sinauer Associates, Sunderland MA, USA) and maximum-likelihood (Tree-Puzzle [Schmidt et al. 2002]) methods.
Results and Discussion
Mining the Human Genome for HERV-K Proviruses
The database of repetitive sequences, Repbase, defines 10 HERV-K families and provides respective consensus sequences. Those different consensus sequences are based on previously available sequence informations and do not necessarily represent optimal sequences when open reading frames, for instance, are of importance. In order to obtain the best possible information on dUTPase sequences within the various HERV-K families, we generated new consensus sequences for the protease domain of each HERV-K family using the human genome sequence. By BLAT-searching the human genome browser (Kent et al. 2002) with Repbase consensus sequences, we identified and subsequently extracted HERV-K proviral sequences. We performed searches for all HERV-K families except HML-2 and HML-3, for which dUTPase consensus sequences have been reported by others and us, respectively (Harris et al. 1997; Mayer and Meese 2002). As shown in Table 1, BLAT searches yielded various numbers of hits and identity scores for each HERV-K family.
Identifying and Collecting HERV-K Family Protease Regions and Searching for dUTPase in Non-HERV-K Families
The previously identified or suggested dUTPase domains for the HERV-K(HML-2) and HML-3 families are located within the N-terminal portion of the protease reading frame (Harris et al. 1997; Mayer and Meese 2002). We reasoned that other HERV-K families harbor potential dUTPase motifs in that region as well. We performed BlastX searches on the Repbase consensus sequences to identify HERV-K regions with protease similarity. From those results we determined a region of about 1 kb for each HERV-K family to harbor protease motifs (Table 1). We selected proviruses containing the respective protease corresponding region or portions thereof by dot matrix comparison.
We could not identify protease motifs in the HERV-K(C4) sequence provided by Repbase. A BlastX search did not detect similarities to proteases. A consensus sequence for the first 4 kb of HERV-K(C4), supposed to contain the protease region, from eight proviral loci displayed two overlapping ORFs, with the N-terminal ORF harboring a Gag_p10 motif and the C-terminal ORF harboring reverse transcriptase motifs. There was no evidence that some HERV-K(C4) proviruses in the human genome contained additional sequence portions within the suspected protease region. We conclude that HERV-K(C4) loci, as they are present in the human genome, lack protease or dUTPase homologous regions. Rather, the gag and polymerase homologous regions seem to overlap each other. Thus, the HERV-K(C4) proviruses that once formed in the genome of the human lineage very likely lacked that proviral region already. We therefore excluded HERV-K(C4) from further analysis.
Furthermore, we did not deduce a dUTPase consensus sequence for HERV-K11DI since only two provirusus harboring the corresponding region were found in the human genome. Both displayed disrupted reading frames. However, three-phase translations subjected to CD-search also revealed clear dUTPase similarities for both proviruses. We therefore conclude the—at that time—presence of dUTPase in HERV-K11DI, for which the exact sequence is uncertain, though.
As protease regions were present in the remaining HERV-K families we extracted corresponding sequences plus about a 250-bp flanking sequence on each side to ensure extraction of the entire protease region. We obtained between 2 and 25 protease sequences for the various HERV-K families (Table 1).
We furthermore searched for dUTPase motifs in HERV families other than HERV-K. To do so, we three-phase translated all HERV internal sequences given in Repbase Update Version 7.8, 54 in total, and subjected the protein sequences to CD-search. While motifs typical for retroviral gag, protease, or polymerase proteins were detected frequently, dUTPase motifs were found only in HERV families for which dUTPase had been reported before. Thus, the various HERV-K families and HERV-L were the only retroviral sequences fixed in the genome of the human evolutionary lineage that harbored dUTPase domains, based on the currently available sequence information.
Generation of Consensus Sequences and ORFs for HERV-K Protease Genes
For each HERV-K family we generated a ClustalW multiple alignment of the extracted sequence portions. The aligned DNA sequences for each HERV-K family displayed clear similarities to each other (Table 1). Consensus sequences for every family were generated employing Boxshade at the Institut Pasteur. We decided ambiguous nucleotide positions according to the majority rule and according to the possible introduction of stop codons. For each family a long central ORF of about 1 kb (948 to 1062 bp) flanked by overlapping ORFs on each side was detected (Table 1). Except for HERV-K(C4) (see above), dUTPase motifs located N-terminal to protease motifs were detected for the central ORF. The upstream-flanking ORFs displayed motifs and similarities typical for retroviral Gag proteins, the downstream flanking ORFs harbored polymerase (reverse transcriptase) motifs. Hence, all newly generated HERV-K consensus sequences displayed a central ORF harboring dUTPase and protease motifs, flanked by overlapping presumable gag and polymerase genes.
Compared to the respective Repbase sequences we found several differences, probably due to a higher number of sequences available at the time of our study (Table 1). A portion of those differences altered the corresponding amino acid and often resulted in the more conserved amino acid. For HERV-K14CI and HERV-K13I our updated sequences corrected one and three frameshifts, respectively, within the dUTPase coding region. In the latter sequence a stop codon was corrected in addition. Further frameshifts and nucleotide differences were revealed when the entire dUTPase/protease gene region was regarded (not shown).
Multiple Alignment of dUTPases
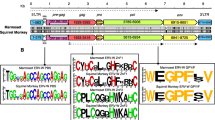
We focused on the dUTPase domain within the protease reading frame. Recent reports assigned the HERV-K(HML-2) dUTPase to a so-called MMTV-related group, including retroviruses such as Mason–Pfizer monkey virus (MPMV), mouse mammary tumor virus (MMTV), and Jaagsiekte sheep retrovirus (JSRV). The so-called nonprimate lentivirus dUTPases, such as feline immunodeficiency virus (FIV), Visna virus, caprine arthritis-encephalitis virus (CAEV), were the most closely related group (Baldo and McClure 1999). We retrieved respective retroviral dUTPase sequences from the pfam dataset for dUTPases and aligned them with the HERV-K dUTPases generated in this study, employing a recently established hidden Markov model for dUTPases (Bateman et al. 2002). As shown in Fig. 1 the HERV-K dUTPases display strong similarities to other retroviral dUTPases. Similarities to MMTV-related sequences were generally higher compared to nonprimate lentiviral sequences.

Multiple alignment of dUTPase sequences employing the hmmalign program and a hidden Markov model for dUTPase. Sequences designated “own” were generated in this study; “rep” sequences were derived from the Repbase consensus sequences. Other dUTPase sequences were obtained from the pfam dUTPase dataset (pfam00692 at http://pfam.wustl.edu ). Note that “own” sequences display differences from the Repbase sequences in several conserved amino acids. Amino acid differences in conserved positions are further specified. Conserved dUTPase motifs 1 to 5 areindicated. CAEV, caprine arthritis encephalitis virus; DUT human, cellular human dUTPase; EIAV, equine infectious anemia virus; JSRV, Jaagsiekte sheep retrovirus; MaeVis, Maedi Visna retrovirus; MarsuEnT-D, marsupial endogenous type D retrovirus; MIAD8, mouse intracisternal A-type particle provirus; MMTV, mouse mammary tumor virus; MoTD, mouse type D endogenous retrovirus; MPMV, Mason–Pfizer monkey virus; Puma/Ovi_Lenti, lentiviruses from puma and cow; SiERVT_D, simian endogenous type D retrovirus; SMRVH, squirrel monkey retrovirus; SRV, simian retrovirus; Visna, Visna virus.
dUTPases usually contain five well-conserved motifs that together comprise the active site of the protein. Amino acids essential for enzymatic function have been described based on sequence alignments of various dUTPases and analysis of crystal structures (Harris et al. 1999; Vassylyev and Morikawa 1996). Most of the non-HERV-K dUTPases shown in Fig. 1 are thought to be active enzymes. Several differences in conserved regions of the alignment were noted (Fig. 1). For amino acid changes in obviously more conserved regions we determined whether the majority of sequences supported a particular amino acid on the DNA level, that is, whether there was evidence for the more conserved amino acid in some sequences. We detected two such instances. As for the M/T difference at amino acid (aa) 38 in HERV-K14I a minority of sequences encoded the conserved threonine (T), while the majority of sequences encoded methionine (M), and a minority of sequences did not support the G/R difference at aa 79 (see above) but, rather, encoded the conserved glycine. In the remaining cases the DNA alignments supported the particular amino acid changes. Based on the consensus sequences generated in this study it cannot be concluded with certainty whether the proviruses that once integrated into, and eventually were fixed in, the germ line encoded a functional dUTPase or had just lost that enzymatic activity. However, our data make it likely that exogenous ancestors of the various HERV-K families indeed encoded functional dUTPase considering (1) the high sequence conservation compared to active dUTPases and (2) the high mutation rate of retroviruses, which probably would have removed a nonfunctional dUTPase from the retroviral genome in a relatively short time period.
Phylogenetic Analysis of HERV-K dUTPases
We analyzed the phylogenetic relationship of the HERV-K dUTPase sequences to other dUTPases by different methods. All methods yielded similar tree topologies and were similar to the results from a previous study (Baldo and McClure 1999). Most HERV-K dUTPase sequences comprised a monophyletic group related to but clearly distinct from the previously defined MMTV relatives (Baldo and McClure 1999). Notably, every method grouped the HERV-K3I and HERV-K22I dUTPase sequences outside the HERV-K clade, and they appear to be more closely related to the MMTV clade than to the remaining HERV-K dUTPases. The HERV-L dUTPase grouped between the MMTV-relatives and the nonprimate lentivirus group (Fig. 2). Thus, most HERV-K families display a common origin when the dUTPase is regarded. Such common origin was also suggested from previous DNA analysis of a conserved polymerase region. Interestingly, the more distant relationship of HERV-K22I/HERV-K(HML-5) and HERV-K3I/HERV-K(HML-6) dUTPases was also indicated by those studies (Medstrand and Blomberg 1993; Tristem 2000).

Phylogenetic tree of dUTPase sequences from HERV-K, MMTV-related, and nonprimate lentiviruses. The tree shown here was generated using PAUP* and represents a minimum evolution consensus tree from 100 bootstraps. The tree was rooted with the human cellular dUTPase sequence as outgroup. Branches with less than 50% bootstrap support were collapsed. The HERV-K Repbase sequences are not included in the tree, as they always grouped nearest to the corresponding HERV-K sequences generated in this study. The MMTV-related clade and the nonprimate lentivirus clade, as recently defined by others (Baldo and McClure 1999), are indicated. For abbreviations see the legend to Fig. 1.
Human endogenous retroviruses are sequence fossils of former exogenous retroviruses that integrated into the germ line genome many million years ago and have remained in the genome since then. Various HERV families underwent sequence changes in the course of continued activity and amplification of proviruses in the host genome following germ line integration, such as HERV-K(HML-2), HERV-K(HML-3), and HERV-H (Mayer and Meese 2002; Mayer et al. 1998; Nelson et al. 1996). However, most sequence features of retroviral genomes remained conserved and sequence motifs for major retroviral proteins are still recognized in many HERV proviral loci. The quality of protein sequences can be improved by generation of consensus sequences that eliminate random nonsense mutations. We recently deduced a fully translatable HERV-K(HML-3) proviral sequence from the otherwise highly mutated proviruses in the human genome (Mayer and Meese 2002). Hence, HERV sequences still hold significant information about exogenous retroviruses having been present many million years ago and the evolution of retroviruses. Here one aspect concerns the invention or occupation of protein domains. As evidenced by the HERV-K(HML-2)-encoded cORF protein (Boese et al. 2001; Magin et al. 1999; Yang et al. 1999), a protein functionally similar to HIVREV (exporting unspliced retroviral RNA from the nucleus) had already been invented at a minimum of 35 million years ago, when exogenous ancestors of HERV-K(HML-2) were present.
Another invention or occupation of a protein domain very likely represents dUTPase that is present in several retroviruses. Recently, horizontal transfer of dUTPase between the MMTV-related and the nonprimate lentiviruses was suggested (Baldo and McClure 1999). Our study corroborates and reveals dUTPase domains in the HERV-K group of endogenous retroviruses that were present as exogenous agents at least 35 million years ago. The currently available data may not allow us to reveal with certainty an ancestral stage of dUTPase and to clarify horizontal transfer events between different clades. Based on our findings the HERV-K3I and HERV-K22I dUTPases may represent an intermediate between the HERV-K and the MMTV-related clades. Further identification of dUTPase harboring endogenous retroviruses in other, nonhuman genomes and regeneration of the ancient dUTPase sequences may eventually give much more detailed insight into the evolution of retroviral dUTPase. The ongoing sequencing projects and the subsequent analysis of the respective genomes’ retroviral content will provide the data for such studies.
References
ML Andersson M Lindeskog P Medstrand B Westley F May J Blomberg (1999) ArticleTitleDiversity of human endogenous retrovirus class II-like sequences. J Gen Virol 80 255–260 Occurrence Handle1:CAS:528:DyaK1MXjs1Khtg%3D%3D Occurrence Handle9934709
AM Baldo MA McClure (1999) ArticleTitleEvolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J Virol 73 7710–7721 Occurrence Handle1:CAS:528:DyaK1MXltlKhtbg%3D Occurrence Handle10438861
A Bateman E Birney L Cerruti R Durbin L Etwiller SR Eddy S Griffiths-Jones KL Howe M Marshall EL Sonnhammer (2002) ArticleTitleThe Pfam protein families database. Nucleic Acids Res 30 276–280 Occurrence Handle1:CAS:528:DC%2BD38Xht12rsro%3D Occurrence Handle11752314
A Boese U Galli M Geyer M Sauter N Mueller-Lantzsch (2001) ArticleTitleThe Rev/Rex homolog HERV-K cORF multimerizes via a C-terminal domain. FEES Lett 493 117–121 Occurrence Handle10.1016/S0014-5793(01)02280-3 Occurrence Handle1:CAS:528:DC%2BD3MXitl2itLg%3D
JM Coffin SH Hughes HE Varmus (1997) Retroviruses. Cold Spring Harbor Laboratory Press Plainview, NY
A Cordonnier JF Casella T Heidmann (1995) ArticleTitleIsolation of novel human endogenous retrovirus-like elements with foamy virus-related pol sequence. J Virol 69 5890–5897 Occurrence Handle1:CAS:528:DyaK2MXns1KhsLc%3D Occurrence Handle7543594
SR Eddy (1998) ArticleTitleProfile hidden Markov models. Bioinformatics 14 755–763 Occurrence Handle10.1093/bioinformatics/14.9.755 Occurrence Handle1:CAS:528:DyaK1MXktlCmtQ%3D%3D Occurrence Handle9918945
JM Harris RH Haynes EM McIntosh (1997) ArticleTitleA consensus sequence for a functional human endogenous retrovirus K (HERV-K) dUTPase. Biochem Cell Biol 75 143–151 Occurrence Handle10.1139/bcb-75-2-143 Occurrence Handle1:CAS:528:DyaK2sXkslWqtr4%3D Occurrence Handle9250362
JM Harris EM McIntosh GE Muscat (1999) ArticleTitleStructure/function analysis of a dUTPase: Catalytic mechanism of a potential chemotherapeutic target. J Mol Biol 288 275–287 Occurrence Handle10.1006/jmbi.1999.2680 Occurrence Handle1:CAS:528:DyaK1MXislWrs7o%3D Occurrence Handle10329142
InstitutionalAuthorNameInternational Human Genome Sequencing Consortium (2001) ArticleTitleInitial sequencing and analysis of the human genome. Nature 409 860–921 Occurrence Handle1:CAS:528:DC%2BD3MXhsFCjtLc%3D Occurrence Handle11237011
J Jurka (2000) ArticleTitleRepbase update: A database and an electronic journal of repetitive elements. Trends Genet 16 418–420 Occurrence Handle1:CAS:528:DC%2BD3cXmvFygtr0%3D
WJ Kent CW Sugnet TS Furey KM Roskin TH Pringle AM Zahler D Haussler (2002) ArticleTitleThe human genome browser at UCSC. Genome Res 12 996–1006 Occurrence Handle1:CAS:528:DC%2BD38Xks12hs7s%3D Occurrence Handle12045153
R Lower J Lower R Kurth (1996) ArticleTitleThe viruses in all of us: Characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci USA 93 5177–5184 Occurrence Handle10.1073/pnas.93.11.5177 Occurrence Handle8643549
C Magin R Lower J Lower (1999) ArticleTitlecORF and RcRE, the Rev/Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J Virol 73 9496–9507 Occurrence Handle1:CAS:528:DyaK1MXmslGqsr4%3D Occurrence Handle10516058
J Mayer E Meese (2002) ArticleTitleThe human endogenous retrovirus family HERV-K(HML-3). Genomics 80 331–343 Occurrence Handle10.1006/geno.2002.6839 Occurrence Handle1:CAS:528:DC%2BD38XmsV2ksb4%3D Occurrence Handle12213204
J Mayer E Meese N Mueller-Lantzsch (1998) ArticleTitleHuman endogenous retrovirus K homologous sequences and their coding capacity in Old World primates. J Virol 72 1870–1875 Occurrence Handle1:CAS:528:DyaK1cXhtFeiu78%3D Occurrence Handle9499038
P Medstrand J Blomberg (1993) ArticleTitleCharacterization of novel reverse transcriptase encoding human endogenous retroviral sequences similar to type A and type B retroviruses: Differential transcription in normal human tissues. J Virol 67 6778–6787 Occurrence Handle1:CAS:528:DyaK2cXisVOitw%3D%3D Occurrence Handle7692084
DT Nelson NL Goodchild DL Mager (1996) ArticleTitleGain of Sp1 sites and loss of repressor sequences associated with a young, transcriptionally active subset of HERV-H endogenous long terminal repeats. Virology 220 213–218 Occurrence Handle10.1006/viro.1996.0303 Occurrence Handle1:CAS:528:DyaK28Xjtl2nt7w%3D Occurrence Handle8659116
M Oliveros R Garcia-Escudero A Alejo E Vinuela ML Salas J Salas (1999) ArticleTitleAfrican swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J Virol 73 8934–8943 Occurrence Handle1:CAS:528:DyaK1MXmslGrtrw%3D Occurrence Handle10515998
K Reus J Mayer M Sauter H Zischler N Mueller-Lantzsch E Meese (2001) ArticleTitleHERV-K(OLD): The ancestor sequences of the human endogenous retrovirus family HERV-K(HML-2). J Virol 75 8917–8926 Occurrence Handle10.1128/JVI.75.19.8917-8926.2001 Occurrence Handle1:CAS:528:DC%2BD3MXntVGrtb8%3D Occurrence Handle11533155
HA Schmidt K Strimmer M Vingron A von Haeseler (2002) ArticleTitleTREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18 502–504 Occurrence Handle10.1093/bioinformatics/18.3.502 Occurrence Handle1:CAS:528:DC%2BD38XivFKrsL0%3D Occurrence Handle11934758
EL Sonnhammer SR Eddy E Birney A Bateman R Durbin (1998) ArticleTitlePfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26 320–322 Occurrence Handle1:CAS:528:DyaK1cXovVChtw%3D%3D Occurrence Handle9399864
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
M Tristem (2000) ArticleTitleIdentification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J Virol 74 3715–3730 Occurrence Handle10.1128/JVI.74.8.3715-3730.2000 Occurrence Handle1:CAS:528:DC%2BD3cXitFOltr4%3D Occurrence Handle10729147
P Turelli G Petursson F Guiguen JF Mornex R Vigne G Querat (1996) ArticleTitleReplication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J Virol 70 1213–1217 Occurrence Handle1:CAS:528:DyaK28Xks1agtg%3D%3D Occurrence Handle8551582
DG Vassylyev K Morikawa (1996) ArticleTitlePrecluding uracil from DNA. Structure 4 1381–1385 Occurrence Handle1:CAS:528:DyaK2sXmsFCh Occurrence Handle8994964
J Yang HP Bogerd S Peng H Wiegand R Truant BR Cullen (1999) ArticleTitleAn ancient family of human endogenous retroviruses encodes a functional homolog of the HIV-1 Rev protein. Proc Natl Acad Sci USA 96 13404–13408 Occurrence Handle10.1073/pnas.96.23.13404 Occurrence Handle1:CAS:528:DyaK1MXns1Gmtbw%3D Occurrence Handle10557333
H Yin P Medstrand A Kristofferson U Dietrich P Aman J Blomberg (1999) ArticleTitleCharacterization of human MMTV-like (HML) elements similar to a sequence that was highly expressed in a human breast cancer: Further definition of the HML-6 group. Virology 256 22–35 Occurrence Handle10.1006/viro.1998.9587 Occurrence Handle1:CAS:528:DyaK1MXhvFWiurw%3D Occurrence Handle10087223
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft to J.M (Ma2298/2-1) and E.M (Me917/16-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mayer, J., Meese, E.U. Presence of dUTPase in the Various Human Endogenous Retrovirus K (HERV-K) Families . J Mol Evol 57, 642–649 (2003). https://doi.org/10.1007/s00239-003-2514-6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2514-6