
Abstract
Nuclear ribosomal DNA (nrDNA) of gymnosperms, especially Pinaceae, is characterized by slow concerted evolution and exhibits substantial ITS-region length variation (975–3663 bp), in sharp contrast to the narrow range (565–700 bp) in angiosperms. Here we examined intra- and intergenomic heterogeneity of the nrDNA internal transcribed spacer (ITS) region in four varieties of Larix potaninii, a species from the mountainous areas of western China. Two clones with more than a 100-bp deletion in ITS1 were detected in L. potaninii var. chinensis and L. potaninii var. australis, respectively. The deletion resulted in the loss of most part, including the motif sequence, of subrepeat 1 (SR1). Sequence divergence and phylogenetic analyses showed that some clones would be pseudogenes given their low GC content, high substitution rates, unique positions in the phylogenetic trees, or significant length variation. These clones might represent orphons or paralogues at minor loci resulting from large-scale gene or chromosome reorganization. Some recombinants characterized by chimeric structure and discordant phylogenetic positions in partitioned sequence analyses indicate that unequal crossing-over plays an important role in the process of nrDNA concerted evolution. In addition, some varieties of L. potaninii might have experienced an nrDNA founder effect parallel to their geographical isolation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nuclear ribosomal DNA (nrDNA) occurs as tandemly repeated units at one or several loci, with copy numbers varying from several hundreds to thousands per haploid genome (Long and Dawid 1980). Each unit, separated by intergenic spacers (IGS), consists of the 18S, 5.8S, and 26S coding regions and two internal transcribed spacers (ITS1 and ITS2) in plants. Repeats within one array are more closely related to one another than to repeats in arrays on other chromosomes (Schlötterer and Tautz 1994; Copenhaver et al. 1995; Muir et al. 2001) or to repeats in other taxa (Arnhein 1983) due to concerted evolution by unequal crossing-over (Smith 1976) and gene conversion (Nagylaki and Petes 1982; Nagylaki 1984; Hillis et al. 1991). For nrDNA of angiosperms, intragenomic diversity is generally low and the length of the ITS region remains in the relatively narrow range of 565 to 700 bp (Baldwin et al. 1995; Wendel et al. 1995). Though some polymorphism was observed within or among individuals, species, or even genera, the ITS repeats do not differ significantly in length, but solely in nucleotide substitutions. The only exception, a variant carrying a 100-bp insertion in ITS1, was reported within and among individuals from five populations of the Lisianthius skinneri species complex of the family Gentianaceae (Sytsma and Schaal 1990).
The concerted evolution of nrDNA in gymnosperms, however, is much slower than that in angiosperms. Substantial length variation is commonly observed. For example, the family Pinaceae exhibits the greatest variation in ITS length, ranging from 1550 bp in Pseudotsuga to between 3150 and 3660 bp in Picea (Liston et al. 1996; Marrocco et al. 1996; Germano and Klein 1999; Maggini et al. 1998, 2000; Gernandt et al. 2001). Besides, inter- or intraspecific length difference in the ITS region as large as several hundred base pairs was reported in Abies (Xiang et al. 2000), Picea (Maggini et al. 1998, 2000), and Pinus (Quijada et al. 1998; Gernandt et al. 2001). Especially, extensive intragenomic length variation (>100 bp) has been found in Cedrus deodara (Liston et al. 1996) and Pinus sylvestris (Karvonen and Savolainen 1993) through RFLP or PCR-RFLP analysis. It is interesting that the large and extensive length variation always results from ITS1, while 5.8S and ITS2 have very conserved lengths, i.e., 162 and 232–245 bp, respectively (Gernandt and Liston 1999; Gernandt et al. 2001). Sequence structure analysis showed that a conserved motif embedded in two to six subrepeats occurs in all Pinaceae ITS1 and is synapomorphic for the family. In particular, Pinaceae ITS1 length variation is highly positively correlated with subrepeat number (Vining 1999). The high heterogeneity of nrDNA in Pinaceae provides us with an ideal material to retrieve the evolutionary process of the multigene family and to explore the mechanism of concerted evolution. However, previous studies focused mainly on two old genera, i.e., Pinus and Picea, with an early Cretaceous origin (Florin 1963; Millar 1993; Wang et al. 2000), and no large intragenomic length variation of the ITS region was found at the sequence level (Bobola et al. 1992; Karvonen and Savolainen 1993; Quijada et al. 1998; Gernandt et al. 2001). In addition, Pinus and Picea have multiple rDNA loci, ranging between five and eight pairs in the diploid genomes (Brown and Carlson 1997; Gernandt and Liston 1999). Some authors suggested that the ITS heterogeneity may be related to the high number of rDNA loci (Liston et al. 1996; Gernandt and Liston 1999; Gernandt et al. 2001). Further studies on the relationship between concerted evolution rate and rDNA loci number are needed.
Contrary to Pinus and Picea, Larix is a young genus with an Eocene origin (LePage and Basinger 1995; Wang et al. 2000) and has fewer rDNA loci (three pairs) (Lubaretz et al. 1996). It consists of about 10 species, which fall into two morphologically distinct groups based on the female cone. Sect. Larix (or Pauciserialis) comprises species characterized by bracts on the cone that did not extend well beyond the seed scales, and Sect. Multiserialis includes species with bracts extending far beyond the seed scales (Patschke 1913; Farjon 1990; LePage and Basinger 1991, 1995; Schorn 1994). Larix potaninii, a species of Sect. Multiserialis, is endemic to the Himalayas and adjacent regions. Due to morphological radiation and geographical isolation, it has differentiated into four varieties, namely, var. potaninii (Pot), var. australis (Aus), var. chinensis (Chi), and var. himalaica (Him) (Farjon 1990, 2001; Fu et al. 1999). The former two varieties are distributed mainly in the Hengduan Mountains, the eastern Himalayan diversity “hot spot” (Wilson 1992), and they are sympatric in SW Sichuan and NW Yunnan. Var. himalaica is confined to the Himalayas (SW Tibet and Nepal), and var. chinensis has a very limited distribution in the Taibai Mountains, S Shaanxi. However, no clear molecular differentiation was found among the four varieties (Wei and Wang 2003). In the present study, we cloned the nrDNA ITS region of all the varieties of L. potaninii to investigate its intra- and intergenomic heterogeneity and to explore the distribution pattern and phylogenetic history of ITS paralogues, which would shed a new light on the evolutionary dynamics of nrDNA family in gymnosperms.
Materials and Methods
Plant Materials
The four varieties of Larix potaninii were all sampled in this study. L. laricina, a North American species of Sect. Larix, was also analyzed. The origins of materials are shown in Table 1.
DNA Extraction, ITS Region Amplification, Cloning, and Sequencing
Total DNA was extracted from silica gel dried needles using the CTAB method following the protocol of Rogers and Bendich (1988) and used as template in the polymerase chain reaction. The ITS region was amplified with primers ITS1N (5′-GTCGTAACAAGGTTTCCGTAGG) located on the 18S rDNA and ITS4 of White et al. (1990). The PCR reaction was carried out in a volume of 25 µl containing 5–50 ng of DNA template, 6.25 pmol of each primer, a 0.2 mM concentration of each dNTP, 2 mM MgCl2, and 0.75 U of Taq DNA polymerase. Amplification was conducted in a Peltier Thermal Cycler (PTC-200, MJ Research). PCR cycles were as follows: 1 cycle of 4 min at 70°C, 4 cycles of 40 s at 94°C, 20 s at 52°C, and 2 min 30 s at 72°C, followed by 36 cycles of 20 s at 94°C, 20 s at 52°C, and 2 min 30 s at 72°C, with a final extension step for 10 min at 72°C. PCR products were separated by 1.5% agarose gel electrophoresis. The band with the right size was cut out and purified using the GFX PCR DNA and Gel Band Purification Kit (Pharmacia) and then cloned with the pGEM-T Easy Vector System II (Promega).
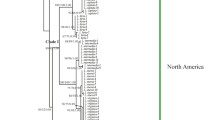
For each taxon, at least 30 clones with correct insertion (determined by digestion with EcoRI) were screened by comparing restriction fragments of MspI or both HaeIII and HinfI. All distinct clones were sequenced with the two PCR primers and several internal primers (Fig. 1), i.e., LITS3 (5′-CTTCTTGCCTCGAGATTTCC), ITS1R1 (5′-CATAACAAGCACACCCATCAC), ITS1R2 (5′-CCTCGTGCAAGACAAAGCAC), and P2N (5′-GAGAGCCGAGATATCCGTTG), using the ABI Prism Bigdye Terminator Cycle Sequencing Ready Reaction Kit. After purification through precipitation with 95% EtOH and 3 M NaAc (pH 5.2), the sequencing reaction products were applied to an ABI 377 automatic sequencer (PE Applied Biosystems, Inc.).

Schematic representation and primer position for ITS region in Larix. Shaded boxes represent the two subrepeats. Horizontal arrows indicate the locations and directions of primers used in the present study. The bases in boldface show the conserved motif sequence in the two subrepeats. The underlined sequences represent an11-bp indel. Numbers below the vertical arrows indicate base position in the aligned ITS data set. The arrowhead represents the core position.
Data Analyses
Boundaries of the ITS, ITS1, 5.8S, and ITS2 regions were identified in comparison with other available sequences (Gernandt and Liston 1999). Sequence alignments were made with CLUSTAL X (Thompson et al. 1997) and refined manually. MEGA v.2.1 (Kumar et al. 2001) was applied to estimate GC content and nucleotide substitutions (d) for the ITS1, 5.8S, and ITS2 regions separately according to Kimura’s (1980) two-parameter model. The standard errors of the estimates were obtained applying 500 bootstrap replicates.
Three data sets—the whole ITS region, 5′ ITS1 partition, and 3′ ITS1+5.8S+ITS2 partition—were analyzed with the maximum parsimony method using PAUP (version 4.0) (Swofford 1998). L. laricina was used as the outgroup considering the female cone morphology (Patschke 1913; Farjon 1990, 2001; Fu et al. 1999; LePage and Basinger 1991, 1995; Schorn 1994) and the congruent result of previous molecular and allozyme locus analyses that Larix was divided into a North American and Eurasian clade (Gernandt and Liston 1999; Semerikov and Lascoux 1999; Wei and Wang 2003). All character states were specified as unordered and equally weighted with indels as missing data. Heuristic search was implemented with 100 random addition sequence replicates, tree bisection–reconnection (TBR) branch swapping, the MULTREES option, and a maximum of 1000 trees saved per round. To evaluate relative robustness of the clades found in the most parsimonious trees, the bootstrap analysis (Felsenstein 1985) employed 100 replicates, with a maximum of 100 trees saved per round using the heuristic search with random sequence addition, TBR branch swapping, and the MULTREES option. The neighbor-joining (NJ) tree was also constructed with MEGA version 2.1 (Kumar et al. 2001) based on the Kimura (1980) two-parameter genetic distance.
Results
Length Heterogeneity and Structure of ITS in Larix potaninii
Twenty-eight distinct clones were recognized by restriction analysis and completely sequenced. The entire ITS region length of the clones ranges from 1653 to 1772 bp. Except Him-22, a clone with a 1-bp deletion in the 5.8S coding region, the others have a conserved length of 5.8S region (162 bp) and ITS2 (232 bp), respectively. Extensive length variation in these clones results from the ITS1 region (Table 2). For example, Aus-17 and Chi-26 have much shorter ITS1, i.e., 1259 and 1260 bp, respectively, than the others (1364–1378 bp). The two clones also have an 11-bp deletion together with Him-5 and Him-22. However, the intragenomic ITS1 length variation was no more than 2 bp for the rest of the clones except Chi-12, with a length of 1364 bp. Generally, except for some unique sequences, the ITS1 region length is relatively conserved in each variety.
Structure analysis showed that the two subrepeats (SR1 and SR2), with a highly conserved center core of GGCCACCCTAGTC embedded, respectively (Gernandt and Liston 1999), also exist in Larix potaninii ITS1 except the two clones Aus-l7 and Chi-26 (Fig. 1). In the alignment matrix of ITS data, SR1 and SR2 covered nucleotide positions 815–878 and 1108–1185 separately, and sequences between position 745 and position 861, including the core of SR1, were deleted for Aus-17 and Chi-26. An 11-bp indel between SR1 and SR2 (positions 1023–1033) was observed in four clones of L. potaninii, namely, Aus-17, Chi-26, Him-5, and Him-22, as well as two clones of L. laricina (Fig. 1). In addition, three GC-rich boxes were detected between nucleotide positions 831–884, 1357–1378, and 1673–1702, with 78, 91, and 77% GC contents, respectively.
GC Content and Sequence Divergence
From Fig. 2 we can see that five clones, Aus-7, Aus-l7, Chi-12, Chi-26, and Him-22, have much more variable nucleotide sites. Therefore, we calculated their GC content and sequence divergence independently. Excluding the five clones, average GC contents of ITS1, 5.8S, and ITS2 in different varieties were 57.0–57.5, 51.3–51.6, and 59.5–59.9%, respectively (Table 2). In general, the GC content in ITS2 was slightly higher than that in ITS1, while that in the 5.8S region was significantly lower than in the two spacers. For the five clones, the GC content in ITS1 (51.3–56.1%) was a little or much lower than the mean value of each taxon. Additionally, Chi-26 had a relatively low GC content (48.1%) in the 5.8S region. In particular, a markedly low GC content was found in each part (ITS1, 5.8S, and ITS2) of the Him-22 ITS region.

Variable nucleotide sites in the aligned ITS sequences of the four varieties of Larix potaninii. Dots indicate identity to the first taxon sequence and indels are indicated as dashes. Shaded sequences denote the unique clones.
Sequence divergence of ITS1, 5.8S, and ITS2 was calculated, respectively, and is shown in Table 3. The five unique clones were not included in the intervarietal divergence calculation in which all clones from the same variety were treated as a group. As a result, great divergence of the whole or partial ITS region was observed among the five clones (not shown) and between the five clones and the four varieties of L. potaninii. Especially, the genetic distance between the three clones, i.e., Chi-26, Aus-17, and Him-22, and the four varieties was two to six times higher than the intervarietal divergence. Surprisingly, Chi-26 and Him-22 showed great divergence of the 5.8S region besides the two spacers. In contrast, Aus-7 and Aus-17 had a low divergence of both the 5.8S region and ITS2. The highest sequence divergence was found in Him-22.
Phylogenetic Analyses
The aligned full-length sequences of the 28 clones from L. potaninii and L. laricina had 1793 characters. Parsimony analysis using heuristic search generated 340 most parsimonious trees with tree length = 379 steps, consistency index = 0.8522, and retention index = 0.7838. One tree that was almost congruent with the strict consensus tree in topology is shown in Fig. 3. A clade comprising Chi-26, Aus-17, and Him-22, three clones with extraordinarily long branch lengths, diverged out first. Aus-7 separated out next. Then the others formed a clade, in which the clones of var. potaninii and var. australis mixed together and formed a monophyletic group (bootstrap value = 70%) as sister to var. chinensis clade. The clones from var. himalaica grouped together as sister clade of the other three varieties. Considering that long-branch attraction is likely to cause phylogenetic errors, we deleted the three clones Chi-26, Him-22, and Aus-17 and reanalyzed the data. The resulting phylogeny was topologically congruent with Fig. 3.

One of the 340 most parsimonious trees constructed from sequence analysis of the ITS region with L. laricina as the outgroup (length = 379, CI = 0.8522, RI = 0.7838). The numbers above and below the branches denote branch lengths and bootstrap percentages greater than 50%, respectively. The numbers following taxa indicate different clones.
For Aus-7, Aus-17, Chi-12, and Chi-26, substitutions were under a great bias toward one of the two ITS fragments separated between SR1 and SR2, and some chimeric structures were clearly observed (Fig. 2). Therefore, the aligned ITS data set was partitioned, between the two subrepeats, into the 5′ITS1 (878 bp) and 3′ITS1+5.8S+ITS2 (915 bp) for further phylogenetic analyses in order to retrieve some recombination events. As expected, the partitioned analyses yielded most parsimonious trees conflicting in topologies, in particular, the positions of the three clones Chi-12, Chi-26, and Aus-7. In the strict consensus tree of 81 most parsimonious trees found in the 5′ITS1 analysis (Fig. 4A), a strongly supported clade, comprising Aus-7, Aus-17, and Him-22, separated out at the base. The other clones from L. potaninii formed a clade, in which three subclades, i.e., var. chinensis, var. himalaica, and var. potaninii + var. australis, were found. Chi-12 and Chi-26 were nested in the var. chinensis subclade. However, in the 3′ITS1 + 5.8S + ITS2 phylogeny (the strict consensus tree of 1000 most parsimonious trees) (Fig. 4B), Chi-26, rather than Aus-7, formed a solidly supported basal clade (87% bootstrap value) together with Aus-17 and Him-22. The clone Chi-12 came out next rather than being nested in var. chinensis subclade. The NJ tree of the complete ITS region was almost same as the MP tree (Fig. 3) in topology. Chi-26, Aus-17, and Him-22 formed a solid clade basal to the other clones of L. potaninii.

Comparison of the strict consensus trees obtained from analyses of partitioned ITS data sets. The clones with gray shading indicate putative recombinants. The numbers above thebranches represent bootstrap values. A 5′ITS1 partition (tree length = 181, CI = 0.8895, RI = 0.8519). B 3′ITS1 + 5.8S + ITS2 partition (tree length = 182, CI = 0.8901,RI = 0.8387).
Discussion
Marked Intragenomic Heterogeneity of ITS and nrDNA Pseudogenes
Attributed to rapid concerted evolution of nrDNA, the ITS paralogues in angiosperms do not differ significantly in length except an intra- and intergenomic length variation of 100 bp in ITS1 documented from the tetraploid Lisianthius skinneri species complex (Gentianaceae) (Sytsma and Schaal 1990). Although interspecific ITS length variation of several hundred base pairs in size has been observed in some taxa of gymnosperms, especially in Pinaceae (Maggini et al. 1998; Xiang et al. 2000; Gernandt et al. 2001), extensive intragenomic length variation of ITS (>100 bp) was reported only in Cedrus deodara (Liston et al. 1996) and 1 of 97 individuals of Pinus sylvestris (Karvonen and Savolainen 1993) through RFLP or PCR-RFLP analysis. In the present study, we found that two divergent clones, Chi-26 and Aus-17, were more than 100 bp shorter than the other clones from the same individual due to a major deletion in the ITS1 region. The deletion resulted in the loss of most of subrepeat 1 (SR1), including the conserved motif; only a very small fragment of SR1 (16 bp) at the 3′ end was retained (Fig. 1). Liston et al. (1996) detected that the ITS1 of Pinaceae is much longer than that of other seed plants and ranges from 1170–1177 bp in Pseudotsuga (Gernandt and Liston 1999) to 2784–3263 bp in Picea (Maggini et al. 2000), which is due in part to the presence of multiple, dispersed subrepeats (Vining 1999). Marrocco et al. (1996) first observed five subrepeats with sequence homology in Pinus pinea, and the sixth subrepeat in this species was reported by Gernandt and Liston (1999). In addition, five to six subrepeats were found in the other species of Pinus (Gernandt et al. 2001), two in Larix (Gernandt and Liston 1999) and three in both Pseudotsuga and Tsuga (Vining and Campbell 1997; Gernandt and Liston 1999). All these subrepeats range from 68 to 265 bp in size with a conserved motif sequence 5′-GGCCACCCTAGTC-3′ (Gernandt and Liston 1999; Vining 1999).
Gernandt and Liston (1999) suggested that subrepeats might play a role in directing rRNA processing considering its high level of sequence conservation and robust behavior in secondary folding models in Larix and Pseudotsuga. If that is true, the loss of most of SR1 in Aus-17 and Chi-26 may imply that the two divergent copies have been released from selective constraint and, therefore, may represent pesudogenes. ITS pseudogenes have been reported in Zea mays (Buckler and Holtsford 1996a, b), Aconitum (Kita and Ito 2000), and Pinus (Gernandt et al. 2001). In particular, two of three divergent rDNA clusters exist as pseudogenes and predate the species divergence of two Quercus species (Muir et al. 2001). These pseudogenes are characterized by an increased rate of substitution or transitional bias, a decreased GC content, a reduction in the spontaneity of secondary structure formation, a decreased number of methylation sites, and a basal or unique position in the phylogenetic trees with long terminal branch lengths. The three clones Aus-17, Chi-26, and Him-22 found in the present study show a clear tendency toward pseudogenes in these characters, especially for Him-22, a unique clone with a markedly low GC content, the greatest sequence divergence, and an extraordinarily long branch length (Table 3, Fig. 3).
ITS Recombinants and Their Implications for Concerted Evolution
nrDNA recombination could generate chimeric molecules, which will most likely be homogenized to a single rDNA molecule, with a sequence intermediate between those of the two ancestral sequences (Muir et al. 2001). The three clones Aus-7, Chi-12, and Chi-26 show great nucleotide substitution bias toward 5′ITS1 or 3′ITS1+5.8S+ITS2 (Fig. 2), and they have discordant positions in phylogenies of the two ITS partitions separated between SR1 and SR2 (Figs. 4A and B), which gives strong evidence for their origin from recombination between divergent nrDNA paralogues. Another clone, Aus-17, shares a major deletion (>100 bp) with Chi-26 besides many synapomorphies between the two clones around the deletion (Figs. 1 and 2), and therefore, it could also be a recombinant sequence. Visual inspection indicated that the unique clones obtained from the same PCR reaction, such as Aus-7 and Aus-17, had different recombination break points and carried specific substitutions. Therefore, they were considered to be true recombinants rather than PCR recombinants (Bradley and Hillis 1997). ITS recombinants have been reported from fungi (Hughes and Petersen 2001) and several angiosperm groups (Buckler et al. 1997; Muir et al. 2001) besides Pinus (Gernandt et al. 2001). Interestingly, an 11-bp indel (Fig. 1) was shared by Chi-26, Aus-17, Him-5, and Him-22 from L. potaninii and all clones of the outgroup L. laricina, a short-bract species endemic to North America, and thus, the indel is very likely characteristic of some ancestral ITS copies of Larix. Besides the synplesiomorphy of the 11-bp indel, Aus-17 and Chi-26 diverged first in the ITS region phylogeny (Fig. 3), implying that the two clones could be evolutionary relicts resulted from recombination among ancient ITS paralogues at nonhomologous loci. In contrast, Aus-7 and Chi-12 are likely to be products of recent recombination events in that they have much shorter branch lengths than Aus-17 and Chi-26 (Figs. 4A and B).
Two mechanisms of recombination commonly invoked as being responsible for concerted evolution are unequal crossing-over (Smith 1976) and gene conversion (Nagylaki and Petes 1982; Nagylaki 1984; Hillis et al. 1991). Gernandt et al. (2001) suggested that the most likely site of recombination, particularly between nonhomologous subrepeats, might be the highly conserved core sequence and that unequal crossing-over resulted in the number variation of tandem subrepeats in Pinaceae. This hypothesis was strongly supported by our finding that most of SR1, including the core, was deleted in the two clones Aus-17 and Chi-26. It is interesting that our data present an apparent paradox: concerted evolution plays an important role in the homogenization of diverse nrDNA repeats, whereas it results in a high level of repeat heterogeneity at the same time. The paradox could be explained by slow concerted evolution in the pine family, which may be attributed to the characteristic genome size, chromosomal organization, and structure of conifer nrDNA repeats and the relatively long generation times (Karvonen et al. 1993; Gernandt et al. 2001).
The number of major 18S–25S rDNA loci in diploid gymnosperm genomes can vary between 3 and 10 (Doudrick et al. 1995; Lubaretz et al. 1996; Brown and Carlson 1997; Tagashira and Kondo 2001; Liu et al. 2003), whereas nonpolyploid angiosperms generally have only 1 or 2 NOR loci (Long and Dawid 1980; Rogers and Bendich 1987), rarely 3 or 4 (Zhang and Sang 1999). The large number of rDNA loci and more subrepeats could slow the rate of repeat homogenization. Moreover, the family Pinaceae is remarkable for its large nuclear genome (Murray 1998) and frequent interspecific hybridization (Carlson and Theroux 1993; Gernandt et al. 2001), whereas it has a highly conserved karyotype (2n = 24, except Pseudolarix), with long metacentric chromosomes of similar size (Brown and Carlson 1997). In order to maintain chromosome stability, divergence and speciation must be accompanied by more complicated chromosome rearrangements in Pinaceae than in angiosperms, which would give rise to some minor loci (Buckler et al. 1997). Liu et al. (2003) observed some weak signals of 18S–25S rDNA loci in some pines using the FISH method and suggested that the weak signals may indicate remnants of primary sites of 18S–25S rDNA that once existed at the centromeres but later moved out to distal sites by chromosome rearrangements. The recombinant clones found in the present study, especially the three putative pseudogenes Aus-17, Chi-26, and Him-22, are likely from the minor loci given their chimeric structure and high sequence divergence. Furthermore, these clones have atypical restriction digestion profiles compared to the other 35 screened clones of respective taxa, a greater AT content and high rate of deamination-type substitutions (Fig. 2, Table 2), indicating that they, particularly Him-22, may exist in only one rDNA locus or represent dispersed solitary genetic elements, that is, orphons (Childs et al. 1981). Karvonen and Savolainen (1993) found that an rDNA variant carrying a 400-bp deletion in ITS1 was present in only one of eight rDNA loci of Pinus sylvestris. Ribosomal DNA (rDNA) orphons have been reported in some angiosperms such as Tripsacum and Winteraceae (Buckler et al. 1997). If the orphon genes or paralogues at minor loci are selectively neutral, they would fasten the divergence and become pseudogenes and, thus, result in the rDNA heterogeneity.
Nevertheless, the concerted evolution rate of the nrDNA family is faster in Larix than in Pinus. All the ITS clones, except some unique ones, show a conserved length and more synapomorphies within each taxon of Larix (Table 2, Fig. 3). This may be attributed to the less complex nrDNA organization, i.e., three NOR loci (Lubaretz et al. 1996), and the short history of Larix (LePage and Basinger 1995; Wang et al. 2000). In contrast, Pinus has an early Cretaceous origin and as many as six to eight NOR loci, which could result in the very slow concerted evolution and extensive intragenomic heterogeneity of nrDNA (Karvonen et al. 1993; Listen et al. 1996; Marrocco et al. 1996; Gernandt et al. 2001). For example, the maximum number of ITS types detected in a single individual was 11, based on 13 clones from P. culminicola (Gernandt et al. 2001).
Founder Effect and Geographical Differentiation of nrDNA in Larix potaninii
As discussed above, extraordinary intra- or intergenomic nrDNA heterogeneity exists in gymnosperms, especially Pinaceae, and has not been completely homogenized by concerted evolution. The nrDNA polymorphisms can disperse among different populations or species through gene flow followed by frequent recombination among nrDNA loci. That is, rDNA paralogues could exist among different species without reproductive isolation. For instance, different ITS clones from the same (or different) individuals of a species were polyphyletic in Pinus subsection Cembroides, a group characterized by frequent interspecific hybridization and very slow concerted evolution (Gernandt et al. 2001). The similar phenomenon was also reported in two closely related Quercus species that frequently hybridize (Muir et al. 2001). Natural hybridization also frequently happens between Larix species, e.g., L. sibirica and L. gmelinii, L. occidentalis and L. lyallii, and L. potaninii and L. mastersiana (Ostenfeld and Larsen 1930; Carlson and Theroux 1993). However, in the present nrDNA phylogeny, all clones, except some unique ones, from var. chinensis and var. himalaica formed a monophyletic group, respectively, though the clones from var. australis and var. potaninii mixed together (Fig. 3), which seems to conflict with the absence of reproductive isolation among closely related larches. The extensive nrDNA differentiation among varieties of L. potaninii could be explained by a founder effect of divergent nrDNA paralogues accompanying geographical isolation.
Var. chinensis is the easternmost variety of L. potaninii and confined to the Qinling Mountains (S Shaanxi), while var. himalaica is the westernmost variety and endemic to the Himalayas. The other two varieties, var. potaninii and var. australis, are distributed in the Hengduan Mountains and geographically located between the former two varieties (Fu et al. 1999; Farjon 2001). The complicated topography of the Himalayan region caused by the uplift of the Tibetan Plateau provided ecological conditions for a nrDNA founder effect in the differentiation process of L. potaninii varieties. Although maximum dispersal distances of the Pinaceae pollen can be 300–1300 km in strong air currents (Potter and Rowley 1960), the nonsaccate pollen (Owens et al. 1998), wet mountainous habitat, and small population size of L. potaninii make long-distance pollen dispersal impossible, which may further result in the fixation and extensive geographical isolation of founder nrDNA copies due to the absence of gene flow among the varieties. A founder effect of multiple gene families has been reported in 5S rDNA of Triticum (Allaby and Brown 2001) and Ophiopogon (Niu and Zhang 2002). As for var. australis and var. potaninii, they are sympatric in SW Sichuan and NW Yunnan, and show only minor differences in the size of the female cone. Therefore, it is not surprising that ITS clones of the two varieties mixed together in the phylogeny.
References
RG Allaby TA Brown (2001) ArticleTitleNetwork analysis provides insights into evolution of 5S rDNA arrays in Triticum and Aegilops. Genetics 157 1331–1341 Occurrence Handle1:CAS:528:DC%2BD3MXisFyktbg%3D Occurrence Handle11238418
N Arnhein (1983) Concerted evolution of multigene families. M Nei RK Koehn (Eds) Evolution of genes and proteins. Sinauer Sunderland MA 38–61
BG Baldwin MJ Sanderson JM Porter MF Wojciechowski CS Campbell MJ Donoghue (1995) ArticleTitleThe ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Ann Mo Bot Gard 82 247–277
MS Bobola DE Smith AS Klein (1992) ArticleTitleFive major nuclear ribosomal repeats represent a large and variable fraction of the genomic DNA of Picea rubens and P. mariana. Mol Biol Evol 9 125–137 Occurrence Handle1:CAS:528:DyaK38XnslKluw%3D%3D Occurrence Handle1552835
RD Bradley DM Hillis (1997) ArticleTitleRecombinant DNA sequences generated by PCR amplification. Mol Biol Evol 14 592–593 Occurrence Handle1:CAS:528:DyaK2sXjtVGntr4%3D Occurrence Handle9159937
GR Brown JE Carlson (1997) ArticleTitleMolecular cytogenetics of the genes encoding 18S-5.8S-26S rRNA and 5S rRNA in two species of spruce (Picea). Theor Appl Genet 95 1–9 Occurrence Handle1:CAS:528:DyaK2sXlvVakt7g%3D
ES Buckler IV TP Holtsford (1996a) ArticleTitle Zea ribosomal repeat evolution and mutation patterns. Mol Biol Evol 13 623–632
ES Buckler IV TP Holtsfo (1996b) ArticleTitle Zea systematics: ribosomal ITS evidence. Mol Biol Evol 13 612–622
ES Buckler A Ippolito TP Holtsford (1997) ArticleTitleThe evolution of ribosomal DNA: Divergent paralogues and phylogenetic implications. Genetics 145 821–832 Occurrence Handle1:CAS:528:DyaK2sXmsVaktLc%3D Occurrence Handle9055091
CE Carlson LE Theroux (1993) ArticleTitleCone and seed morphology of western larch (Larix occidentalis), alpine larch (Larix lyallii), and their hybrids. Can J For Res 21 1264–1269
G Childs R Maxson RH Cohn L Kedes (1981) ArticleTitleOrphons: Dispersed genetic elements derived from tandem repetitive genes of Eucaryotes. Cell 23 651–663 Occurrence Handle1:CAS:528:DyaL3MXhs12ltLY%3D Occurrence Handle6784929
GP Copenhaver JH Doelling JS Gens CS Pikaard (1995) ArticleTitleUse of RFLPs larger than 100-kb to map the position and internal organization of the nucleolus organizer region on chromosome-2 in Arabidopsis thaliana. Plant J 7 273–286 Occurrence Handle10.1046/j.1365-313X.1995.7020273.x Occurrence Handle1:CAS:528:DyaK2MXkslGntLw%3D
RL Doudrick JS Heslop-Harrison CD Nelson T Schmidt WL Nance T Schwarzacher (1995) ArticleTitleKaryotype of slash pine (Pinus elliottii var. elliottii) using patterns of fluorescence in situ hybridization and fluorochrome banding. J Hered 86 289–296
A Farjon (1990) Pinaceae. Koeltz Scientific Books Konigstein, Germany
A Farjon (2001) World checklist and bibliography of conifers, 2nd ed. Royal Botanic Gardens Kew, UK
J Felsenstein (1985) ArticleTitleConfidence limits on phylogenies: An approach using the bootstrap. Evolution 39 783–791
R Florin (1963) ArticleTitleThe distribution of conifers and taxad genera in time and space. Acta Hort Berg 20 121–312
L-K Fu N Li RR Mill (1999) Larix. Z-Y Wu PH Raven (Eds) Flora of China (4). Science Press, Beijing, and Missouri Botanical Garden Press St. Louis 33–37
J Germane AS Klein (1999) ArticleTitleSpecies-specific nuclear and chloroplast single nucleotide polymorphisms to distinguish Picea glauca, P. mariana and P. rubens. Theor Appl Genet 99 37–49 Occurrence Handle10.1007/s001220051206 Occurrence Handle1:CAS:528:DyaK1MXlvFGlt7o%3D
DS Gernandt A Liston (1999) ArticleTitleInternal transcribed spacer region evolution in Larix and Pseudotsuga (Pinaceae). Am J Bot 86 711–723 Occurrence Handle1:CAS:528:DyaK1MXjslersr0%3D Occurrence Handle10330075
DS Gernandt A Liston D Piñero (2001) ArticleTitleVariation in the nrDNA ITS of Pinus subsection Cembroides: Implications for molecular systematic studies of pine species complexes. Mol Phylogenet Evol 21 449–467 Occurrence Handle10.1006/mpev.2001.1026 Occurrence Handle1:CAS:528:DC%2BD3MXptVWhsbs%3D Occurrence Handle11741386
DM Hillis C Moritz CA Porter RJ Baker (1991) ArticleTitleEvidence for biased gene conversion in concerted evolution of ribosomal DNA. Science 251 308–310 Occurrence Handle1:CAS:528:DyaK3MXps1WjsQ%3D%3D Occurrence Handle1987647
KW Hughes RH Petersen (2001) ArticleTitleApparent recombination or gene conversion in the ribosomal ITS region of a flammulina (Fungi, Agaricales) hybrid. Mol Biol Evol 18 94–96 Occurrence Handle1:CAS:528:DC%2BD3MXhtVSmsL8%3D Occurrence Handle11141197
P Karvonen M Karjalainen O Savolainen (1993) ArticleTitleRibosomal RNA genes in Scots pine (Pinus sylvestris L.): Chromosomal organization and structure. Genetica 88 59–68 Occurrence Handle1:CAS:528:DyaK2cXpslKrsQ%3D%3D
P Karvonen O Savolainen (1993) ArticleTitleVariation and inheritance of ribosomal DNA in Pinus sylvestris L. (Scots pine). Heredity 71 614–622 Occurrence Handle1:CAS:528:DyaK2cXitFaksbw%3D
M Kimura (1980) ArticleTitleA simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16 111–120 Occurrence Handle7463489
Y Kita M Ito (2000) ArticleTitleNuclear ribosomal ITS sequences and phylogeny in East Asian Aconitum subgenus Aconitum (Ranunculaceae), with special reference to extensive polymorphism in individual plants. Plant Syst Evol 225 1–13 Occurrence Handle1:CAS:528:DC%2BD3MXhsFGjsrk%3D
S Kumar K Tamura IB Jakobsen M Nei (2001) MEGA2: Molecular Evolutionary Genetics Analysis software. Arizona State University Tempe
BA LePage JF Basinger (1991) ArticleTitleA new species of Larix (Pinaceae) from the early Tertiary of Axel Heiberg Island, Arctic Canada. Rev Palaeobot Palyno 70 89–111 Occurrence Handle10.1016/0034-6667(91)90080-M
BA LePage JF Basinger (1995) ArticleTitleThe evolutionary history of the genus Larix (Pinaceae). USDA For Serv Int Res Sta GTR-INT 319 19–29
A Liston WA Robinson JM Oliphant (1996) ArticleTitleLength variation in the nuclear ribosomal DNA internal transcribed spacer region of non-flowering seed plants. Syst Bot 21 109–120
Z-L Liu D Zhang X-R Wang (2003) ArticleTitleChromosomal localization of 5S and 18S-5.8S-25S ribosomal DNA sites in five Asian pines using fluorescence in situ hybridization. Theor Appl Genet 106 198–204 Occurrence Handle1:CAS:528:DC%2BD3sXltlehtQ%3D%3D Occurrence Handle12582844
EO Long IB Dawid (1980) ArticleTitleRepeated genes in eukaryotes. Annu Rev Biochem 49 727–764 Occurrence Handle1:CAS:528:DyaL3cXltV2jt7s%3D Occurrence Handle6996571
O Lubaretz J Fuchs R Ahne A Meister I Schubert (1996) ArticleTitleKaryotyping of three Pinaceae species via fluorescent in situ hybridization and computer-aided chromosome analysis. Theor Appl Genet 92 411–416 Occurrence Handle10.1007/s001220050143
F Maggini R Marrocco MT Gelati RI De Dominicis (1998) ArticleTitleLengths and nucleotide sequences of the internal spacers of nuclear ribosomal DNA in gymnosperms and pteridophytes. Plant Syst Evol 213 199–205
F Maggini M Frediani MT Gelati (2000) ArticleTitleNucleotide sequence of the internal transcribed spacers of ribosomal DNA in Picea abies Karst. DNA Seq 11 87–89 Occurrence Handle1:CAS:528:DC%2BD3MXhtVGru7s%3D Occurrence Handle10902913
R Marrocco MT Gelati F Maggini (1996) ArticleTitleNucleotide sequence of the internal transcribed spacers and 5.8S region of ribosomal DNA in Pinus pinea L. DNA Seq 6 175–177 Occurrence Handle1:CAS:528:DyaK2sXotFOrsg%3D%3D Occurrence Handle8722573
CI Millar (1993) ArticleTitleImpact of the Eocene on the evolution of Pinus L. Ann Mo Bot Gard 80 471–498
G Muir CC Fleming C Schlotterer (2001) ArticleTitleThree divergent rDNA clusters predate the species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol Biol Evol 18 112–119 Occurrence Handle1:CAS:528:DC%2BD3MXotVOmsg%3D%3D Occurrence Handle11158370
BG Murray (1998) ArticleTitleNuclear DNA amounts in gymnosperms. Ann Bot 82 IssueIDSuppl A 3–15 Occurrence Handle1:CAS:528:DyaK1MXkt1Wksg%3D%3D
T Nagylaki (1984) ArticleTitleEvolution of multigene families under interchromosomal gene conversion. Proc Natl Acad Sci USA 81 3769–3800
T Nagylaki TD Petes (1982) ArticleTitleIntrachromosomal gene conversion and the maintenance of sequence homogeneity among repeated genes. Genetics 100 315–337 Occurrence Handle1:CAS:528:DyaL38Xlt1Wgt70%3D Occurrence Handle7106560
W Niu D Zhang (2002) ArticleTitleEvolution of 5S rRNA genes in Ophiopogon xylorrhizus Wang et Dai and O. sylvicola Wang et Tang (Convallariaceae). Acta Bot Sin 44 329–336 Occurrence Handle1:CAS:528:DC%2BD38XktVaksbo%3D
CH Ostenfeld CS Larsen (1930) ArticleTitleThe species of the genus Larix and their geographic distribution. Biol Meddel Kongel Danske Vidensk Selsk 9 1–107
JN Owens T Takaso CJ Runions (1998) ArticleTitlePollination in conifers. Trends Plant Sci 3 479–485 Occurrence Handle10.1016/S1360-1385(98)01337-5
W Patschke (1913) ArticleTitleÜber die extratropischen ostasiatischen Coniferen und ihre Bedeutung für die pflanzengeographische Gliederung Ostasiens. Bot Jahrb Syst 48 626–776
LD Potter J Rowley (1960) ArticleTitlePollen rain and vegetation, San Augustin Plains, New Mexico. Bot Gaz 122 1–25 Occurrence Handle10.1086/336081
A Quijada A Liston P Delgado A Vazquez-Lobo ER Alvarez-Buylla (1998) ArticleTitleVariation in the nuclear ribosomal DNA internal transcribed spacer (ITS) region of Pinus rzedowskii revealed by PCR-RFLP. Theor Appl Genet 96 539–544 Occurrence Handle10.1007/s001220050771 Occurrence Handle1:CAS:528:DyaK1cXivVWlsLo%3D
SO Rogers AJ Bendich (1987) ArticleTitleRibosomal RNA genes in plants: Variability in copy number and in the intergenic spacer. Plant Mol Biol 9 509–520 Occurrence Handle1:CAS:528:DyaL2sXmtFartbw%3D
SO Rogers AJ Bendich (1988) ArticleTitleExtraction of DNA from plant tissues. Plant Mol Biol Manual A6 1–10
C Schlötterer D Tautz (1994) ArticleTitleChromosomal homogeneity and Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Curr Biol 47 777–783
HE Schorn (1994) ArticleTitleA preliminary discussion of fossil larches (Larix, Pinaceae) from the Arctic. Quat Internal 22/23 173–183 Occurrence Handle10.1016/1040-6182(94)90011-6
VL Semerikov M Lascoux (1999) ArticleTitleGenetic relationship among Eurasian and American Larix species based on allozymes. Heredity 83 62–70 Occurrence Handle10.1038/sj.hdy.6885310 Occurrence Handle1:CAS:528:DyaK1MXlsFygu7Y%3D Occurrence Handle10447704
GP Smith (1976) ArticleTitleEvolution of repeated DNA sequences by unequal crossover. Science 191 528–535 Occurrence Handle1:CAS:528:DyaE28Xhs1Srurg%3D Occurrence Handle1251186
DL Swofford (1998) PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4. Sinauer Sunderland, MA
KJ Sytsma BA Schaal (1990) ArticleTitleRibosomal DNA variation within and among individuals of Lisianthius (Gentianaceae) populations. Plant Syst Evol 170 97–106 Occurrence Handle1:CAS:528:DyaK3MXjs1ygsQ%3D%3D
N Tagashira K Kondo (2001) ArticleTitleChromosome phylogeny of Zamia and Ceratozamia by means of Robertsonian changes detected by fluorescence in situ hybridization (FISH) technique of rDNA. Plant Syst Evol 227 145–155 Occurrence Handle1:CAS:528:DC%2BD3MXms1ehs78%3D
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleCLUSTAL-X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25 4876–4882 Occurrence Handle1:CAS:528:DyaK1cXntFyntQ%3D%3D Occurrence Handle9396791
Vining TF (1999) Molecular phylogenetics of Pinaceae. PhD dissertation. University of Maine, Orono
TF Vining CS Campbell (1997) ArticleTitlePhylogenetic signal in sequence repeats within nuclear ribosomal DNA internal transcribed spacer 1 in Tsuga. Am J Bot 84 IssueIDSuppl 702
X-Q Wang DC Tank T Sang (2000) ArticleTitlePhylogeny and divergence times in Pinaceae: evidence from three genomes. Mol Biol Evol 17 773–781 Occurrence Handle1:CAS:528:DC%2BD3cXjt1Gjtb8%3D Occurrence Handle10779538
X-X Wei X-Q Wang (2003) ArticleTitlePhylogenetic split of Larix: Evidence from paternally inherited cpDNA trnT-trnF region. Plant Syst Evol 239:67–77 239 67–77 Occurrence Handle10.1007/s00606-002-0264-3
JF Wendel A Schnabel T Seelanan (1995) ArticleTitleBidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci USA 92 280–284 Occurrence Handle1:CAS:528:DyaK2MXjtVens7o%3D Occurrence Handle7816833
TJT White SL Bruns J Taylor (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. M Innis D Gelfand J Sninsky T White (Eds) PCR protocols: A guide to methods and applications. Academic Press San Diego 315–322
EO Wilson (1992) The diversity of life. Belknap Press of Harvard University Press Cambridge, MA
Q-P Xiang Q-Y Xiang A Listen L-K Fu D-Z Fu (2000) ArticleTitleLength variation of the nuclear ribosomal DNA internal transcribed spacer in the genus Abies, with reference to its systematic utility in Pinaceae. Acta Bot Sin 42 946–951 Occurrence Handle1:CAS:528:DC%2BD3cXotVamurg%3D
D Zhang T Sang (1999) ArticleTitlePhysical mapping of ribosomal RNA genes in peonies (Paeonia, Paeoniaceae) by fluorescent in situ hybridization: Implications for phylogeny and concerted evolution. Am J Bot 86 735–740 Occurrence Handle1:CAS:528:DyaK1MXjslersrs%3D Occurrence Handle10330077
Acknowledgements
We thank Drs. Zhou Shi-Liang and Ding Kai-Yu for help in field collection and Miss Sun Ying-Xue for help in sequencing. This work was supported by grants from the State Key Basic Research and Development Plan (Grant G2000046804) and Chinese Academy of Sciences (Grant kscxz-sw-101A and the special fund to Wang Xiao-Quan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wei, XX., Wang, XQ. & Hong, DY. Marked Intragenomic Heterogeneity and Geographical Differentiation of nrDNA ITS in Larix potaninii (Pinaceae) . J Mol Evol 57, 623–635 (2003). https://doi.org/10.1007/s00239-003-2512-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2512-8