
Abstract
The evolution of viral genomes has recently attracted considerable attention. We compare the sequences of two large viral genomes, EsV-1 and FirrV-1, belonging to the family of phaeoviruses which infect different species of marine brown algae. Although their genomes differ substantially in size, these viruses share similar morphologies and similar latent infection cycles. In fact, sequence comparisons show that the viruses have more than 60% of their genes in common. However, the order of genes is completely different in the two genomes, suggesting that extensive recombinational events in addition to several large deletions had occurred during the separate evolutionary routes from a common ancestor. We investigated genes encoding components of signal transduction pathways and genes encoding replicative functions in more detail. We found that the two genomes possess different, although overlapping, sets of genes in both classes, suggesting that different genes from each class were lost, perhaps randomly, after the separate evolution from an ancestral genome. Random loss would also account for the fact that more than one-third of the genes in one viral genome has no counterparts in the other genome. We speculate that the ancestral genome belonged to a cellular organism that had once invaded a primordial brown algal host.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Viruses constitute a heterogeneous collection of biological agents whose common characteristic is that they depend on metabolic functions of their cellular hosts for multiplication. This basic tenet is realized in nature by a bewildering variety of concepts. The evolution of viruses is a fascinating field of research that has recently attracted considerable attention (Gibbs et al. 1995; Strauss and Strauss 2001; Iyer et al. 2001; Bell 2001; Takemura 2001; Lawrence et al. 2002; Hendrix 2002). A reasonable assumption is that viruses with either RNA or DNA genomes, and possibly even distinct groups of RNA- or DNA-containing viruses, arose by different evolutionary routes.
Here we consider a class of viruses with very large and complex DNA genomes—phaeoviruses that infect marine brown algal species (phaeos, Gr. brown). Phaeoviruses, together with the related but distinct chloroviruses (infecting Chlorella species), are members of the phycodnavirus family and share icosahedral morphologies with internal lipid membranes and large double-stranded DNA genomes (Müller and Knippers 2001). Specific phaeoviruses are named according to their host species in which they occur (for a review see Müller et al. 1998). All presently known phaeoviruses infect free-swimming, wall-less gametes or spores. The viral DNA becomes integrated into the cellular genome and is then transmitted via mitosis through all cell generations of the developing host. The viral genome remains latent in vegetative cells and is only expressed in cells of the reproductive algal organs, sporangia and gametangia. Here the viral genome is massively replicated until the cellular organelles break down and become replaced by densely packed viral particles. Virions are released into the surrounding seawater under stimuli such as changes in light and temperature that also trigger the release of spores and gametes. Thus, free virions appear in the same environment in synchrony with susceptible host cells and, therefore, guarantee the initiation of new infection cycles. Except for partial or total inhibition of reproduction, infected algae show no apparent growth or developmental defects, and phaeoviruses are pandemic in several brown algal species examined (Müller et al. 1998).
Most phaeoviruses have DNA genomes in the 335-kb size range like EsV-1, the Ectocarpus siliculosus virus-1, whose nucleotide sequence has recently been described (Delaroque et al. 2001). Exceptions are viruses of the brown algal genus Feldmannia. The Feldmannia sp. virus, (FsV), infects an unidentified Feldmannia species. It has genomes of 160–180 kb, although its multiplication cycle is similar to that of EsV-1 and other phaeoviruses (Henry and Meints 1992; Lee et al. 1995, 1998a,b; Ivey et al. 1996; Krueger et al. 1996). Here we describe a second Feldmannia virus, FirrV-1, which is endemic in the species Feldmannia irregularis (Müller and Frenzer 1993). The DNA in FirrV-1 virions consists of a linear DNA molecule of about 180 kb in addition to many smaller DNA fragments of lengths between 170 and 10 kb (Fig. 1).

Pulsed field gel electrophoresis of viral DNA. Virus particles were embedded in 1% low-melting point agarose and digested with proteinase K. Electrophoresis was carried out in an 0.8% agarose with 45 mM Tris–borate–EDTA buffer (pH 8.0) using a CHEF mapper system (Bio-Rad) at 14°C for 21 h (voltage gradient, 6.0 V·cm−1; included angle,120°; linear switch time ramp, 5.5–36.5 s). Lane M, DNA size markers (concatemeric lambda DNA); lane 1, Ectocarpus siliculosus virus-1 (EsV-1); lane 2, Feldmannia irregularis virus-1 (FirrV-1).
In order to compare the FirrV-1 genome with that of EsV-1 we have sequenced a total of more than 190 kb of the FirrV-1 genome. We could identify 156 open reading frames (ORFs), 93 of which are structurally related to genes in the EsV-1 genome (Delaroque et al. 2001). This high degree of sequence similarity is not surprising given the close taxonomic relationship between the two brown algal hosts, Ectocarpus and Feldmannia, and the highly similar viral infection cycles. However, we wondered why EsV-1 needs a 335-kb-genome with 231 genes to support a life style very similar to that of FirrV-1 with its much smaller genome. Our comparison of the two genomes showed that EsV-1 harbors a number of genes with apparently similar, possibly redundant, functions, whereas the FirrV-1 genome has a much lower number of functionally related genes. Based on these and other observations, we conclude that features of the extant viral genomes can be best explained by assuming that they independently evolved by a loss of genes from a common ancestor (“regression” [Luria and Darnell 1967]). The surprising complexity of phaeoviral genomes is also interesting with respect to recent arguments suggesting that a complex DNA virus could be the “ancestor” of the eukaryotic nucleus, and therefore constitute the basis for “eukaryogenesis” (Bell 2001).
Materials and Methods
Genomic Sequencing of FirrV-1
The cultivation of algae, isolation and purification of viruses, and pulsed field gel electrophoresis technique have been previously described (Lanka et al. 1993; Kapp et al. 1997).
A FirrV-1 shotgun library was constructed according to the procedure used for the EsV-1 genomic library (Delaroque et al. 2001). DNA sequencing was carried out with the Big Dye kit (Perkin–Elmer) according to Sanger et al. (1977) and sequences were analyzed on an ABI 377 sequencer (GATC Biotech, Konstanz, Germany). The shotgun sequences were assembled with the SeqManll Lasergene Software (DNASTAR Inc.) until a coverage between 4 and 8 was reached. PCR was used to close gaps between DNA segments, resulting in 16 contigs.
Analysis of Sequence Data
The ORFs were identified with the lasergene biocomputing software package (DNASTAR Inc.) according to the same criteria used for defining the EsV-1 ORFs (Delaroque et al. 2001). Homology searches were carried out with the Blast program (Altschul et al. [1990] scoring matrix blosum 62). Protein motifs were searched against the SMART (Letunic et al. 2002), PROSITE (Falquet et al. 2002), and Pfam (Bateman et al. 2002) databases. Charged amino acid clusters were found with the SAPS program (Brendel et al. 1992). Sequence alignments were performed with the Megalign program (DNASTAR Inc.). A detailed analysis of the FirrV-1 genes is available as supplementary material from N. Delaroque.
Nucleotide Sequence Accession Numbers
The DNA nucleotide sequences of the FirrV-1 contigs have been deposited in GenBank under the following accession numbers: AY225133 (contig A), AY225134 (contig B), AY225135 (contig C), AY225136 (contig D), AY225137 (contig E), AY225138 (contig F), AY225139 (contig G), AY225140 (contig H), AY225141 (contig I), AY225142 (contig J), AY225143 (contig K), AY225144 (contig L), AY225145 (contig M), AY225146 (contig N), AY225147 (contig O), and AY225148 (contig P).
Results and Discussion
Comparing Genomes
Phaeoviruses infecting the filamentous brown algal hosts Ectocarpus siliculosus and Feldmannia irregularis have similar electron microscopic structures and indistinguishable lysogenic infection cycles. Thus, it could be assumed that these viruses possess genomes of similar sizes. However, the genome of EsV-1 is a continuous DNA molecule of 335 kb with complementary ends that anneal to form a circular molecule (Lanka et al. 1993; Delaroque et al. 2001), whereas the genomes of the two known Feldmannia viruses, FsV (Henry and Meints 1992; Ivey et al. 1996) and FirrV-1, are much smaller. FirrV-1 DNA, prepared from virions, consists of a linear molecule with an electrophoretic length of about 180 kb and a spectrum of DNA fragments in the range of 10–170 kb (Fig. 1).
To understand their genetic relationship better, we decided to determine the sequence of the FirrV-1 genome and to compare it with the published EsV-1 sequence (Delaroque et al. 2001). The total length of the sequenced FirrV-1 DNA was 191,667 bp, which was distributed over several contigs. Numerous PCR assays were performed to close the existing gaps in the sequence, but a contiguous sequence corresponding to the electrophoretically determined 180-kb viral genome could not be obtained. A reason for this may be that DNA fragments were much more abundant than the 180-kb DNA molecule in our preparations of virion DNA (Fig. 1). Since each one of the sequences was determined several times (coverage: 4–8), it can be concluded that the total 190 kb of sequenced DNA contains most and probably all FirrV-1 genes.
The experimentally determined FirrV-1 contigs are ordered in Fig. 2 according to their sizes (A through P; see supplementary material online for details). Surprisingly, several small contigs (I, J, K, L, N, O, P) have sequences that are similar to regions in the largest contigs, A and B (Fig. 2). These regions contain the same genes in the same order but have only between 50 and 80% codons in common (see supplementary material online). It is thus likely that a given FirrV-1 preparation includes virions whose genomes deviate to a certain extent from the majority type. Heterogeneity of viral genomes has also been described for the related Feldmannia virus FsV-1 (Henry and Meints 1992; Lee et al. 1995, 1998a,b; Ivey et al. 1996; Krueger et al. 1996) but has never been encountered in EsV-1 DNA preparations.

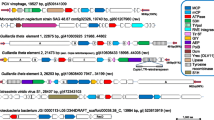
Gene maps. Comparison of the EsV-1 genome (taken from Delaroque et al. 2001) with the sequenced FirrV-1 genomic regions and the three published FsV DNA segments (Krueger et al. 1996; Lee et al. 1998a,b). The DNA sequences are shown as horizontal lines, where thick rectangles represent regions of repetitive elements (labeled A through I in the EsV-1 genome) and triangles are the inverted repeats in the EsV-1 genome. Nucleotide coordinates of EsV-1 are shown below its map. The lengths of contiguous DNA sequences are indicated by the numbers (bp) in parentheses. Each ORF is shown as a vertical bar. ORFs transcribed in the rightward direction are located above the lines and ORFs transcribed leftward are below. Short vertical bars represent ORFs specific for a particular viral genome, and long bars show orthologous ORFs (connected by thin lines). Genes with similar functions are colored according to the key. Major common genes are indicated below the EsV-1 genome by thin arrows.
A characteristic feature of the EsV-1 genome is several large blocks of repetitive elements (Delaroque et al. 2001), whereas FirrV-1 DNA appears to have only a few regions with sequence repeats (Fig. 2), although it cannot be excluded that the unclonable parts of FirrV-1 DNA between contigs consist of repetitive DNA.
We have searched for ORFs according to previously established criteria (Schuster et al. 1990; Delaroque et al. 2001). We identified 156 FirrV-1 genes that are almost equally distributed between the two complementary DNA strands (Fig. 2). The majority of the FirrV-1 genes (93, or 60%) are structurally related to EsV-1 genes, as expected for viruses that have very similar multiplication cycles in taxonomically related host organisms. Surprisingly, however, the order of the related genes is completely different in the two viral genomes (Fig. 2), indicating that extensive recombinations must have occurred since the divergence from a common ancestor.
The sequences of three short regions of another Feldmannia virus. FsV, have been published (Lee et al. 1995, 1998a,b; Krueger et al. 1996). The gene order in two of these regions is exactly as in the FirrV-1 genome, and the third sequenced FsV region contains one deviation from the FirrV-1 gene order.
In spite of extensive gene shuffling, genes shared by EsV-1 and FirrV-1 are not randomly distributed over their respective genomes. Most noticeably, the leftmost 50 kb-region of the EsV-1 genome up to repeat element C entirely lacks FirrV-1 gene orthologues (Fig. 2). This particular region must have been lost during the evolution of FirrV-1, possibly because the 25 EsV-genes in this region were dispensable for viral multiplication in the Feldmannia host.
This may also be the case for a second genomic region on EsV-1, between repeat elements F and G (nucleotide coordinates, 260 and 290 kb; Fig. 2), that lacks FirrV-1 orthologues. A third EsV-1 genomic region without FirrV-1 orthologues resembles a transposon that is bracketed by inverted repeats which both contain genes encoding IS4-like transposases (nucleotide coordinates, 198–223 kb; Fig. 2) (Delaroque et al. 2001). While the putative transposon with its inverted repeats does not occur in the FirrV-1 genome, several of the enclosed genes are conserved and have spread to other parts of the FirrV-1 DNA.
The three EsV-1 genome segments without FirrV-1 orthologues together encompass about 110 kb of DNA and contain many genes that are similar to genes elsewhere on the EsV-1 genome (see supplementary material online). It is therefore possible that the genomic regions, present in EsV-1 but absent in FirrV-1, contain redundant genes. This would, at least partially, explain why FirrV-1 with a genome size of only 180–190 kb is able to maintain a lifestyle indistinguishable from that of EsV-1 with its 335-kb genome.
In summary, almost two-thirds of the EsV-1 and FirrV-1 genes are structurally related and have similar sequences. This is a strong argument that the two viral genomes share a common ancestor, but the different order of the conserved genes indicates that extensive gene shuffling occurred during their separate evolutionary routes. Gene shuffling could have selective advantages as suggested by Zhang et al. (2002). This process leads to rapid phenotypic improvement in bacteria. Thus, gene shuffling may be the result not simply of random recombinatorial processes, but of selection as well, and a phaeovirus which invades a new algal species may benefit from rearranging its gene order.
Comparing Genes
As mentioned above, EsV-1 and FirrV-1 have 93 related genes in common. Most of these genes share between 31 and 50% of their codons, and only 15 genes have more than 50% identical codons (Fig. 3; see also supplementary material online). These 15 genes could be particularly relevant for phaeoviral replication. However, nine of the most highly conserved genes encode proteins of unknown functions, one gene (ORF F2 of FirrV-1) may encode a lipase, and four genes have the potential to code for proteins involved in DNA metabolism including the small and the large subunit of ribonucleotide reductase (A19 and A20), a helicase-like ATPase (B27), and the viral processivity factor PCNA (A6). These genes are considered in more detail below. Finally, a FirrV-1 gene (B50) with particularly high similarity (73%) to an EsV-1 orthologue (ORF 116) could encode a major capsid protein. Iyer et al. (2001) have found related genes in each of four diverse classes of viruses with large DNA genomes. A related gene has not been found (and arguably does not exist) in cellular genomes. Therefore, a comparison of the known capsid gene sequences could illustrate the evolutionary relationship between viruses with large DNA genomes. Not unexpectedly, phylogenetic analyses show that the capsid genes of the two phaeoviruses, EsV-1 and FirrV-1, are closely related to, but equally distant from, the biologically similar Chlorella virus (another phycodnavirus) and to the animal viruses with large genomes (not shown) (Iyer et al. 2001).

FirrV-1 genes and their similarities to EsV-1 genes. The number of ORFs in the FirrV-1 genome is plotted versus the percentage of identical codons.
Next we compare two sets of genes with related functions because they well illustrate the divergent evolution of the two phaeoviral species. We choose for comparisons the genes of hybrid histidine kinases involved in signal transduction pathways and the genes for DNA replication.
Signal Transduction
It was a surprising discovery that phaeoviruses possess genes with the potential to code for hybrid histidine kinases (Delaroque et al. 2000). Hybrid histidine kinases are members of a large family of two-component systems that function in stimulus–response transduction in bacteria, archaea, yeasts, and plants but not in mammalian cells. The main function of two-component systems is to sense changes in the environment and to induce the appropriate genetic response. Most cells express a dozen or more different hybrid kinases (Stock et al. 2000; West and Stock 2001). While widespread in bacteria and lower eukaryotes, (hybrid) histidine kinases have not been found before in other viral systems and appear to be a unique feature of phaeoviruses.
The EsV-1 genome has no fewer than six genes for hybrid histidine kinases and an additional gene for a phosphoshuttle protein that is also a constituent of a two-component signal transduction pathway. As described in detail before, the sequences reveal features of prokaryotic and eukaryotic enzymes (Delaroque et al. 2000, 2001). In contrast, FirrV-1 has only three hybrid histidine kinase genes plus one gene for the phosphoshuttle protein (Fig. 4A). This gene and two of the hybrid kinase genes are conserved between the two viruses including a kinase (EsV-1 ORF 181) with a phytochrome chromophore-binding domain as found in corresponding enzymes from plants and bacteria (Delaroque et al. 2001) (Fig. 4B). However, the third FirrV-1 hybrid kinase gene has no counterpart in the EsV-1 genome.

Phaeoviral hybrid kinases and phosphoshuttle. A List of ORFs encoding the putative hybrid kinases and a phosphoshuttle protein present in EsV-1 and FirrV-1. B Comparison of the putative chromophore binding sites of ORF 181 (EsV-1) and ORF H1 (FirrV-1) with corresponding domains of plant phytochrome and bacteriophytochromes. Arabidopsis thaliana phytochrome E (PHYE; GenBank accession number P42498): Synechocystis sp. Cph1 (SyCph1; GenBank accession number Q55168); Deinococcus radiodurans BphP (DrBphP; GenBank accession number Q9RZA4). Asterisks: cysteine and histidine to which the chromophore is attached in the plant and bacterial phytochrome domains. C Scheme indicating the possible evolution of EsV-1 and FirrV-1 from the common ancestor based on the phaeoviral hybrid histidine kinase and phosphoshuttle genes. The numbered blocks correspond to the ORFs listed in A. Shaded boxes: ORFs shared by EsV-1 and FirrV-1.
While the two-component signal transduction systems may have important (although unknown) functions in the phaeoviral life cycle, it is not at all obvious why EsV-1 needs seven, but FirrV-1 only four genes, of which one is not related to any one of the EsV-1 genes. A possible interpretation is that the two viruses evolved from a common ancestor which had at least eight histidine kinase genes, of which four were deleted during FirrV-1 evolution and one during EsV-1 evolution (Fig. 4C). Thus, with respect to the hybrid kinase genes, the extant viral genomes could be “frozen” states of evolutionary processes (Gray et al. 1999).
We note that, in addition to hybrid histidine kinases, phaeoviruses also have the potential to code for another signal transduction system including a threonine/serine protein kinase. The corresponding gene is conserved between EsV-1 (Delaroque et al. 2001) and FirrV-1 (supplementary material online) and has also been detected in the related Feldmannia virus FsV (Lee et al. 1998a).
DNA Replication
Even though phycodnaviruses replicate in the nuclei of infected cells like other large DNA viruses, they do not entirely depend on cellular functions for DNA replication but encode some of their own replicative enzymes.
A minimal set of replicative functions includes initiator proteins and enzymes for double-strand unwinding and for DNA synthesis (Table 1). Initiator proteins recognize the start points of replication (origins) and induce an initial unwinding of the bound DNA duplex. Well-characterized viral initiator proteins such as the large T antigen of simian virus 40 (Simmons 2000) or the E1 and E2 proteins of papilloma viruses (Lambert 1991), have no sequence similarities to known cellular functions. This could also be true for phaeoviral initiator proteins and may explain why data bank searches gave no information as to which of the phaeoviral sequences could encode an initiator protein.
In contrast, genes for other replicative proteins could be readily identified as shown in Table 1. We include in this comparison not only the two phaeoviruses, but also the Chlorella virus PBCV-1, a distantly related phycodnavirus (Van Etten and Meints 1999), because this gives interesting insights into the replication machines of large viral genomes.
Each of the three phycodnaviruses is capable of expressing a DNA polymerase with a 3′–5′ proofreading exonuclease domain. The phycodnaviral DNA polymerases have sequence similarities to the B family of eukaryotic delta-type DNA polymerases (Hübscher et al. 2002). Villareal and DeFillippis (2000) have placed phycodnaviral polymerase sequences near the root of a clade containing all eukaryotic delta-type DNA polymerases and concluded that the phaeoviral polymerases were close to the evolutionary basis from which this type of eukaryotic DNA polymerases evolved.
DNA polymerases require a primase for the synthesis of RNA primers. Indeed, the three sequenced phycodnaviral genomes possess a gene related to bacteriophage genes of the Siphoviridae family encoding a primase/helicase belonging to the superfamily III of helicases (Gorbalenya et al. 1990). The primase/helicase sequences of the three phycodnaviral genes share between 30 and 50% of their codons with each other and about 25% with the Siphoviridae genes but have no apparent similarity to the bacterial primase (DnaG of E. coli) or the eukaryotic primase (DNA polymerase α-primase).
RNA primers must be removed before strand ligation. A possible candidate for a phycodnaviral RNase could be encoded by FirrV-1 gene A3 and its orthologues in EsV-1 and in PBCV-1 (Table 1).
Delta-like DNA polymerases require the sliding clamp protein PCNA for processive DNA synthesis. Interestingly, the two sequenced phaeoviral genomes contain one gene, and the Chlorella virus genome two genes for PCNA (Table 1). Processivity factors like PCNA must constantly be loaded during discontinuous DNA synthesis on the lagging DNA strand. The eukaryotic pentameric loading complex is known as replication factor C (RFC) and is composed of one large and four different small subunits. Interestingly, only EsV-1 encodes all five RFC subunits, whereas FirrV-1 and the Chlorella virus each possess a gene for the large subunit only. Surprisingly, the large phycodnaviral RFC subunit resembles an archaeal RFC-like protein (Pisani et al. 2000; Bohlke et al. 2002), whereas the four small RFC subunits of EsV-1 have similarities to eukaryotic proteins (Mossi and Hübscher 1998).
Other essential replicative functions include a DNA ligase, a topoisomerase, and a single-strand specific binding (SSB) protein. Indeed, the Chlorella virus, but not EsV-1 and FirrV-1, has genes with the potential to code for a ligase and a topoisomerase (Table 1). However, a candidate gene for a SSB protein has not yet been identified in the phycodnaviral genomes.
Thus, the Chlorella virus encodes a most complete set of replicative functions with the exception of the small RFC subunits, whereas EsV-1 has the capacity for the expression of the entire pentameric RFC protein but lacks genes for a ligase and a topoisomerase, similar to FirrV-1, which, however, also lacks genes for the small RFC subunits (Table 1).
Why do the three related phycodnaviruses encode different components of the replication apparatus? A parsimonious explanation is that a common ancestor possessed all genes required for DNA replication and that differential gene loss occurred during the separate evolution of the extant phycodnaviral genomes. This could also be the case for genes involved in nucleotide metabolism. The Chlorella virus genome has more than a dozen genes with possible functions for the synthesis of DNA precursors (Van Etten and Meints 1999). Only a few of these genes are also found in the two phaeoviral genomes. An example is a FirrV-1 gene for a deoxycytidine deaminase (ORF A29), which is, however, not present in the EsV-1 genome. On the other hand, genes for the large and the small subunit of ribonucleotide reductase are present in the Chlorella virus genome as well as in both phaeoviral genomes (and several other large DNA viruses) (Blasco 1995). It is remarkable that these two genes are highly conserved, while most other genes for deoxynucleotide synthesis were lost. An explanation would be that each viral type requires a specific combination of virally and of cellularly encoded replication functions. However, we see no biochemical reason for this and find it more plausible to assume that the extant viral genomes evolved from a larger ancestor genome (with a full complement of replication genes) by gene losses of different degrees.
We note, finally, that a distinguishing feature of phaeoviruses is a long latency period when the viral DNA is integrated into the host genome and transmitted from cell to cell during development (Müller 1991; Bräutigam et al. 1995; Delaroque et al. 1999). Consequently, the phaeoviruses EsV-1 and FirrV-1 encode a protein with similarities to the catalytic domain of the bacteriophage-type integrase family of site-specific recombinases (Nunes-Düby et al. 1998). Another phaeoviral gene with putative functions in integration shares similarity with the bacteriophage N15 protelomerase (Deneke et al. 2000, 2002; Ravin et al. 2002). This enzyme is known to convert circular into linear DNA, and vice versa. It is significant that genes for an integrase and a protelomerase are not present in the Chlorella virus genome. They are not needed because of the strictly lytic infection cycle of the Chlorella virus.
Conclusions
Genome sequences of three phycodnaviruses are now known, namely, that of the chlorovirus PBCV-1 (Van Etten and Meints 1999) and those of two phaeoviruses, EsV-1 (Delaroque et al. 2001) and FirrV-1 (this work). In addition, several DNA short sequences of the phaeovirus FsV have been described (Lee et al. 1995, 1998 a,b; Krueger et al. 1996).
First inspection reveals that these viruses are related because they share a significant number of orthologous genes (Van Etten et al. 2002). It is also clear that the two phaeoviruses have more genes in common and are therefore more closely related to each other than each one is to PBCV-1. This is expected as PBCV-1 infects Chlorella and initiates a lytic infection cycle, whereas EsV-1 and FirrV-1 evolved to perform lysogenic (latent) infections in taxonomically related marine brown algal hosts. The genes, shared by EsV-1 and FirrV-1, must include those that are essential for the phaeoviral infection cycle. An example is the integrase gene, which is probably required for the integration of viral DNA into the host genome as a condition for the establishment of latency.
However, each virus has an additional number of genes without counterparts in the related virus. In fact, the EsV-1 genome contains about 75 more genes than the FirrV-1 genome, and well over one-third of the FirrV-1 genes have no orthologues in the EsV-1 genome.
This can be formally explained by one of two models. The first model (“gain of genes”) assumes that each evolving virus acquired specific sets of genes. The genes may be necessary for an optimal multiplication in the particular host organism. The mechanisms could be multiple lateral gene transfers, and indeed, phycodnaviral genomes contain elements resembling eubacterial, archaeal, and eukaryotic genes (Van Etten et al. 2002).
The second model (“loss of genes”) proposes that the two viral genomes are descendants of a common ancestor. The large ancestral genome is thought to include all the genes shared by the two viruses in addition to the genes which are specific for each virus. Accordingly, the common ancestor, perhaps a single-cell organism, invaded and may have first lived in symbiosis with a primordial brown algal host and later shed more and more of its genes when it began to adopt a viral lifestyle in the evolving algal hosts. As outlined above, this model explains best why the two viral genomes contain different, although overlapping sets of hybrid kinase genes (see Fig. 4) and why they have different combinations of replication genes (see Table 1). We presently prefer this second model and conclude that an original cellular ancestor evolved by gene loss, recombinations, and other mechanisms into the extant viruses with their unusually complex genomes. This does not necessarily imply that viral evolution led linearly from the cellular ancestor to present-day viruses. Instead, the evolution of phaeoviruses could have been “modular” (Botstein 1980), and gene loss could be accompanied by the acquisition of gene segments from various sources via lateral gene transfer. An example of an acquired gene may be the major capsid gene (ORF 116 in EsV-1, B50 in FirrV-1), of which homologues are found in all large DNA viruses but not in cellular organisms (Iyer et al. 2001). The proposal that some viruses may have evolved from a symbiotic cellular organism is not a new idea in virology (see: Blasco 1995). We believe, however, that particularly clear traces of the process of genomic simplification or “regression” can be found in the genomes of extant phaeoviruses.
References
SF Altschul W Gish W Miller EW Myers DJ Lipman (1990) ArticleTitleBasic local alignment search tool. J Mol Biol 215 403–410 Occurrence Handle10.1006/jmbi.1990.9999 Occurrence Handle1:CAS:528:DyaK3MXitVGmsA%3D%3D Occurrence Handle2231712
A Bateman E Birney L Cerruti R Durbin L Etwiller SR Eddy S Griffiths-Jones KL Howe M Marshall EL Sonnhammer (2002) ArticleTitleThe Pfam protein families database. Nucleic Acids Res 30 276–280 Occurrence Handle1:CAS:528:DC%2BD38Xht12rsro%3D Occurrence Handle11752314
PJ Bell (2001) ArticleTitleViral eukaryogenesis: Was the ancestor of the nucleus a complex DNA virus? J Mol Evol 53 251–256 Occurrence Handle10.1007/s002390010215 Occurrence Handle1:CAS:528:DC%2BD3MXnt1Ghs7c%3D Occurrence Handle11523012
R Blasco (1995) Evolution of poxviruses and African swine fever virus. AJ Gibbs CH Calisher F García-Arenal (Eds) Molecular basis of virus evolution. Cambridge University Press Cambridge, UK 255–268
K Bohlke FM Pisani M Rossi G Antranikian (2002) ArticleTitleArchaeal DNA replication: spotlight on a rapidly moving field. Extremophiles 6 1–14 Occurrence Handle10.1007/s007920100222 Occurrence Handle1:CAS:528:DC%2BD38XitVKnu78%3D Occurrence Handle11878556
D Botstein (1980) ArticleTitleA theory of modular evolution for bacteriophages. Ann NY Acad Sci 354 484–490 Occurrence Handle1:CAS:528:DyaL3MXpvFKktQ%3D%3D Occurrence Handle6452848
M Bräutigam M Klein R Knippers DG Müller (1995) ArticleTitleInheritance and meiotic elimination of a virus genome in the host Ectocarpus siliculosus (Phaeophyceae). J Phycol 31 823–827
V Brendel P Bucher I Nourbakhsh BE Blaisdell S Karlin (1992) ArticleTitleMethods and algorithms for statistical analysis of protein sequences. Proc Natl Acad Sci USA 89 2002–2006 Occurrence Handle1:CAS:528:DyaK38Xkt1arsrY%3D Occurrence Handle1549558
N Delaroque I Maier R Knippers DG Müller (1999) ArticleTitlePersistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J Gen Virol 80 1367–1370 Occurrence Handle1:CAS:528:DyaK1MXjs1OmtLg%3D Occurrence Handle10374952
N Delaroque S Wolf DG Müller R Knippers (2000) ArticleTitleThe brown algal virus EsV-1 particle contains a putative hybrid histidine kinase. Virology 273 383–390 Occurrence Handle10.1006/viro.2000.0383 Occurrence Handle1:CAS:528:DC%2BD3cXltFemtb0%3D Occurrence Handle10915609
N Delaroque DG Müller G Bothe T Pohl R Knippers W Boland (2001) ArticleTitleThe complete DNA sequence of the Ectocarpus siliculosus virus EsV-1 genome. Virology 287 112–132 Occurrence Handle10.1006/viro.2001.1028 Occurrence Handle1:CAS:528:DC%2BD3MXlvFWit7o%3D Occurrence Handle11504547
J Deneke G Ziegelin R Lurz E Lanka (2000) ArticleTitleThe protelomerase of temperate Escherichia coli phage N15 has cleaving-joining activity. Proc Natl Acad Sci USA 97 7721–7726 Occurrence Handle10.1073/pnas.97.14.7721 Occurrence Handle1:CAS:528:DC%2BD3cXkvFCnt7w%3D Occurrence Handle10884403
J Deneke G Ziegelin R Lurz E Lanka (2002) ArticleTitlePhage N15 telomere resolution. Target requirements for recognition and processing by the protelomerase. J Biol Chem 277 10410–10419 Occurrence Handle10.1074/jbc.M111769200 Occurrence Handle1:CAS:528:DC%2BD38XisVylu7Y%3D Occurrence Handle11788606
L Falquet M Pagni P Bucher N Hulo CJ Sigrist K Hofmann A Bairoch (2002) ArticleTitleThe PROSITE database, its status in 2002. Nucleic Acids Res 30 235–238 Occurrence Handle1:CAS:528:DC%2BD38Xht12ku70%3D Occurrence Handle11752303
AJ Gibbs CH Calisher F García-Arenal (Eds) (1995) Molecular basis of virus evolution. Cambridge University Press Cambridge
AE Gorbalenya EV Koonin YI Wolf (1990) ArticleTitleA new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett 262 145–148 Occurrence Handle10.1016/0014-5793(90)80175-I Occurrence Handle1:CAS:528:DyaK3cXitFalu7w%3D Occurrence Handle2156730
MW Gray G Burger BF Lang (1999) ArticleTitleMitochondrial evolution. Science 283 1476–1481 Occurrence Handle1:CAS:528:DyaK1MXhs1ygt70%3D Occurrence Handle10066161
RW Hendrix (2002) ArticleTitleBacteriophages: Evolution of the majority. Theor Popul Biol 61 471–480 Occurrence Handle10.1006/tpbi.2002.1590 Occurrence Handle12167366
EC Henry RH Meints (1992) ArticleTitleA persistent virus infection in Feldmannia (Phaeophyceae). J Phycol 28 517–526
U Hübscher G Maga S Spadari (2002) ArticleTitleEukaryotic DNA polymerases. Annu Rev Biochem 71 133–163
RG Ivey EC Henry AM Lee L Klepper SK Krueger RH Meints (1996) ArticleTitleA Feldmannia algal virus has two genome size-classes. Virology 220 267–273 Occurrence Handle10.1006/viro.1996.0314 Occurrence Handle1:CAS:528:DyaK28Xjs1ymt74%3D Occurrence Handle8661377
LM Iyer L Aravind EV Koonin (2001) ArticleTitleCommon origin of four diverse families of large eukaryotic DNA viruses. J Virol 75 11720–11734 Occurrence Handle10.1128/JVI.75.23.11720-11734.2001 Occurrence Handle1:CAS:528:DC%2BD3MXotlaqs7c%3D Occurrence Handle11689653
M Kapp R Knippers DG Müller (1997) ArticleTitleNew members of a group of DNA viruses infecting brown algae. Phycol Res 45 85–90 Occurrence Handle1:CAS:528:DyaK2sXktlOrt70%3D
SK Krueger RG Ivey EC Henry RH Meints (1996) ArticleTitleA brown algal virus genome contains a “RING” zinc finger motif. Virology 219 301–303 Occurrence Handle10.1006/viro.1996.0251 Occurrence Handle1:CAS:528:DyaK28XislCnsLY%3D Occurrence Handle8623545
PF Lambert (1991) ArticleTitlePapillomavirus DNA replication. J Virol 65 3417–3420 Occurrence Handle1:CAS:528:DyaK3MXksVaku7Y%3D Occurrence Handle1645776
ST Lanka M Klein U Ramsperger DG Müller R Knippers (1993) ArticleTitleGenome structure of a virus infecting the marine brown alga Ectocarpus siliculosus. Virology 193 802–811 Occurrence Handle10.1006/viro.1993.1189 Occurrence Handle1:CAS:528:DyaK3sXit1Ohsbo%3D Occurrence Handle8460486
JG Lawrence GF Hatfull RW Hendrix (2002) ArticleTitleThe imbroglios of viral taxonomy: Genetic exchange and the failings of phenetic approaches. J Bacteriol 184 4891–4905 Occurrence Handle10.1128/JB.184.17.4891-4905.2002 Occurrence Handle1:CAS:528:DC%2BD38Xmt12ns74%3D Occurrence Handle12169615
AM Lee RG Ivey EC Henry RH Meints (1995) ArticleTitleCharacterization of a repetitive DNA element in a brown algal virus. Virology 212 474–480 Occurrence Handle10.1006/viro.1995.1505 Occurrence Handle1:CAS:528:DyaK2MXosFWgtr4%3D Occurrence Handle7571417
AM Lee RG Ivey RH Meints (1998a) ArticleTitleRepetitive DNA insertion in a protein kinase ORF of a latent FsV (Feldmannia sp. Virus) genome. Virology 248 35–45 Occurrence Handle1:CAS:528:DyaK1cXlsFOlsbs%3D
AM Lee RG Ivey RH Meints (1998b) ArticleTitleThe DNA polymerase gene of a brown algal virus: Structure and phylogeny. J Phycol 34 608–615 Occurrence Handle1:CAS:528:DyaK1cXmtFKnsL0%3D
I Letunic L Goodstadt NJ Dickens T Doerks J Schultz R Mott F Ciccarelli RR Copley CP Ponting P Bork (2002) ArticleTitleRecent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res 30 242–244 Occurrence Handle10.1093/nar/30.1.242 Occurrence Handle1:CAS:528:DC%2BD38Xht12ku7s%3D Occurrence Handle11752305
SE Luria JE Darnell (1968) General virology, 2nd ed. Wiley London
R Mossi U Hübscher (1998) ArticleTitleClamping down on clamps and clamp loaders. The eukaryotic replication factor C. Eur J Biochem 254 209–216 Occurrence Handle1:CAS:528:DyaK1cXjvFeitL4%3D Occurrence Handle9660172
DG Müller (1991) ArticleTitleMendelian segregation of a virus genome during host meiosis in the marine brown alga Ectocarpus siliculosus. J Plant Physiol 137 739–743
DG Müller K Frenzer (1993) ArticleTitleVirus infections in three marine brown algae: Feldmannia irregularis, F. simplex, and Ectocarpus siliculosus. Hydrobiologia 260/261 37–44
DG Müller R Knippers (2001) Phaeovirus, Phycodnaviridae. CA Tidona G Darai (Eds) The springer index of viruses, an authoritative reference source of virology. Spinger-Verlag Berlin 732–736
DG Müller M Kapp R Knippers (1998) ArticleTitleViruses in marine brown algae. Adv Virus Res 50 49–67 Occurrence Handle9520996
SE Nunes-Düby HJ Kwon RS Tirumalai T Ellenberger A Landy (1998) ArticleTitleSimilarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26 391–406 Occurrence Handle9421491
FM Pisani M De Felice F Carpentieri M Rossi (2000) ArticleTitleBiochemical characterization of a clamp-loader complex homologous to eukaryotic replication factor C from the hyperthermophilic archaeon Sulfolobus solfataricus. J Mol Biol 301 61–73 Occurrence Handle10.1006/jmbi.2000.3964 Occurrence Handle1:CAS:528:DC%2BD3cXltlCjs78%3D Occurrence Handle10926493
NV Ravin TS Strakhova VV Kuprianov (2001) ArticleTitleThe protelomerase of the phage-plasmid N15 is responsible for its maintenance in linear form. J Mol Biol 312 899–906 Occurrence Handle10.1006/jmbi.2001.5019 Occurrence Handle1:CAS:528:DC%2BD3MXntFKqtLY%3D Occurrence Handle11580235
F Sanger S Nicklen AR Coulsen (1977) ArticleTitleDNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci USA 74 5463–5467 Occurrence Handle271968
AM Schuster M Graves K Korth M Ziegelbein J Brumbaugh D Grone RH Meints (1990) ArticleTitleTranscription studies of a 4.3-kbp fragment from a ds-DNA eukaryotic algal virus. Virology 176 515–523 Occurrence Handle1:CAS:528:DyaK3cXltFSju70%3D Occurrence Handle2345963
DT Simmons (2000) ArticleTitleSV40 large T antigen functions in DNA replication and transformation. Adv Virus Res 55 75–134 Occurrence Handle1:CAS:528:DC%2BD3cXosV2qs7k%3D Occurrence Handle11050941
AM Stock VL Robinson PN Goudreau (2000) ArticleTitleTwo-component signal transduction. Annu Rev Biochem 69 183–215
JH Strauss EG Strauss (2001) ArticleTitleVirus evolution: how does an enveloped virus make a regular structure? Cell 105 5–8 Occurrence Handle1:CAS:528:DC%2BD3MXivFOjsb0%3D Occurrence Handle11300997
M Takemura (2001) ArticleTitlePoxviruses and the origin of the eukaryotic nucleus. J Mol Evol 52 419–425 Occurrence Handle1:CAS:528:DC%2BD3MXksVGnurs%3D Occurrence Handle11443345
JL Van Etten RH Meints (1999) ArticleTitleGiant viruses infecting algae. Annu Rev Microbiol 53 447–494 Occurrence Handle10.1146/annurev.micro.53.1.447 Occurrence Handle1:CAS:528:DyaK1MXmvVOnsbo%3D Occurrence Handle10547698
JL Van Etten MV Graves DG Muller W Boland N Delaroque (2002) ArticleTitlePhycodnaviridae-large DNA algal viruses. Arch Virol 147 1479–1516 Occurrence Handle10.1007/s00705-002-0822-6 Occurrence Handle1:CAS:528:DC%2BD38XovVamur0%3D Occurrence Handle12181671
LP Villarreal VR DeFilippis (2000) ArticleTitleA hypothesis for DNA viruses as the origin of eukaryotic replication proteins. J Virol 74 7079–7084 Occurrence Handle10.1128/JVI.74.15.7079-7084.2000 Occurrence Handle1:CAS:528:DC%2BD3cXkvFCmtbc%3D Occurrence Handle10888648
AH West AM Stock (2001) ArticleTitleHistidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 26 369–376 Occurrence Handle10.1016/S0968-0004(01)01852-7 Occurrence Handle1:CAS:528:DC%2BD3MXkt1Kht78%3D Occurrence Handle11406410
YX Zhang K Perry VA Vinci K Powell WP Stemmer SB del Cardayre (2002) ArticleTitleGenome shuffling leads to rapid phenotypic improvement in bacteria. Nature 415 644–646 Occurrence Handle10.1038/415644a Occurrence Handle1:CAS:528:DC%2BD38XhsVCjsbk%3D Occurrence Handle11832946
Acknowledgements
We thank Eric B. Carstens for comments and suggestions and the GATC team for their cooperation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Delaroque, N., Boland, W., Gerhard Müller, D. et al. Comparisons of Two Large Phaeoviral Genomes and Evolutionary Implications . J Mol Evol 57, 613–622 (2003). https://doi.org/10.1007/s00239-003-2501-y
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2501-y