
Abstract
Carboxylesterases hydrolyze esters of short-chain fatty acids and have roles in animals ranging from signal transduction to xenobiotic detoxification. In plants, however, little is known of their roles. We have systematically mined the genome from the model plant Arabidopsis thaliana for carboxylesterase genes and studied their distribution in the genome and expression profile across a range of tissues. Twenty carboxylesterase genes (AtCXE) were identified. The AtCXE family shares conserved sequence motifs and secondary structure characteristics with carboxylesterases and other members of the larger α/β hydrolase fold superfamily of enzymes. Phylogenetic analysis of the AtCXE genes together with other plant carboxylesterases distinguishes seven distinct clades, with an Arabidopsis thaliana gene represented in six of the seven clades. The AtCXE genes are widely distributed across the genome (present in four of five chromosomes), with the exception of three clusters of tandemly duplicated genes. Of the interchromosomal duplication events, two have been mediated through newly identified partial chromosomal duplication events that also include other genes surrounding the AtCXE loci. Eighteen of the 20 AtCXE genes are expressed over a broad range of tissues, while the remaining 2 (unrelated) genes are expressed only in the flowers and siliques. Finally, hypotheses for the functional roles of the AtCXE family members are presented based on the phylogenetic relationships with other plant carboxylesterases of known function, their expression profile, and knowledge of likely esterase substrates found in plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Carboxylesterases (EC 3.1.1.1) are a large family of enzymes that hydrolyze esters of short-chain fatty acids. Structurally carboxylesterases belong to the α/β hydrolase fold superfamily of proteins (Ollis et al. 1992). Members of the α/β hydrolase superfamily contain a conserved structural core consisting of eight β-strands, with connecting α-helices and loops interspersed within and around the eight-β-strand core. The main feature of carboxylesterases is the conserved catalytic triad. The active site is made up of a serine (surrounded by the conserved consensus sequence G–X–S–X–G), a glutamate (or less frequently an aspartate), and a histidine (Ollis et al. 1992; Oakeshott et al. 1999). These residues are dispersed throughout the primary amino acid sequence but come together in the tertiary structure to form a charge relay system, creating a nucleophilic serine that can attack the substrate. Another structural motif of importance is the oxyanion hole, which is involved in stabilizing the substrate–enzyme intermediate during hydrolysis. The oxyanion hole is created by three small amino acids: two glycine residues typically located between β-strand 3 and α-helix 1 and the third located immediately following the catalytic serine residue (Ollis et al. 1992).
Carboxylesterases have a broad range of functions including neurotransmission in animals (Taylor and Radic 1994), pheromone degradation in moths (Vogt et al. 1985), reproductive fitness in flies (Oakeshott et al. 1987), degradation of xenobiotics in mammalian livers (reviewed by Satoh and Hosokawa 1998), insecticide resistance in insects (Hemingway 2000), and xenobiotic detoxification in microbes (Turner et al. 2002). Some carboxylesterases are highly specific, acting on only a particular substrate at very high rates (e.g., acetylcholine [Sussman et al. 1991]), while others are able to hydrolyze a broad range of substrates (e.g., Oakeshott et al. 1999).
While the carboxylesterases have been intensively studied in animals (Satoh and Hosokawa 1998; Oakeshott et al. 1999), they have not been well characterized in plants. Their use as isoenzyme markers has demonstrated that multiple carboxylesterases are present in many plants and that they are expressed in many tissues, including leaves, fruit, and roots (e.g., apple [Manganaris and Alston 1992]; barley [Kahler and Allard 1970], cherry [Boskovic and Tobutt 1998], maize, [MacDonald and Brewbaker 1974], pear [Fachinello et al. 2000], tomato [Tanksley and Rick 1980], and yams [Dansi et al. 2000]). These isoenzymes studies have also allowed estimation of the number of loci that encode carboxylesterases: 4 loci in apple (Manganaris and Alston 1992), 10 loci from barley (Kahler and Allard 1970), 9 loci in cherry (Boskovic and Tobutt 1998), and 10 loci each from maize (MacDonald and Brewbaker 1974) and tomato (Tanksley and Rick 1980). Despite this work, there are few reports indicating a physiological role for these enzymes.
Five carboxylesterase genes have been isolated from plants. They all share conserved structural motifs and the catalytic triad characteristic for the carboxylesterase family. Four of these genes have a role in plant–pathogen interactions. In Nicotiana tabacum, hsr203J is induced during the hypersensitive response to attempted infection by the bacterium, Ralstonia solanacearum (Pontier et al. 1994). Antisense experiments in N. tabacum demonstrate a role for hsr203J in suppressing programmed cell death associated with the hypersensitive response during pathogen attack (Tronchet et al. 2001). A Lycopersicon esculentum ortholog of the hsr203J gene, Lehsr203J, is also upregulated in response to a pathogen, in this case the AVR9 peptide from the fungus Cladosporium fulvum (Pontier et al. 1998). In addition, inoculation of Pisum sativum with the fungus Mycosphaerella pinodes causes an increase in the expression of the hsr203J ortholog, E86 (Ichinose et al. 2001). PepEST from Capsicum annuum is another carboxylesterase involved in plant–pathogen interactions; however, it is not orthologous to the three hsr203J genes (Kim et al. 2001). Increased expression of PepEST is observed during infection of pepper plants with the necrotrophic pathogen, Colletotrichum gloeosporioides (Kim et al. 2001). Application of recombinantly produced PepEST onto C. annuum fruit was found to confer a weak level of resistance to pathogen attack (Kim et al. 2001). The fifth plant carboxylesterase gene PrMC3, has been isolated from Pinus radiata (Walden et al. 1999). It is expressed in male reproductive structures but, as yet, has no known physiological role.
Given the variety of physiological roles attributed to carboxylesterases identified from animals and microbes, a wider number of roles for carboxylesterases in plants outside those identified to date is likely. As a prelude to determining their function in plants, here we describe the carboxylesterase gene family from the model plant, Arabidopsis thaliana (L.) Heynh (Arabidopsis). We characterize the 20 Arabidopsis carboxylesterase (AtCXE) genes, detailing their phylogenetic associations, genomic organization, and broad tissue expression patterns.
Materials and Methods
Bioinformatics
Five previously published plant carboxylesterase genes were retrieved from GenBank and used to identify the AtCXE genes in the Arabidopsis genome (The Arabidopsis Genome Initiative 2000) at the TAIR web site (Huala et al. 2001) by key word and iterative TBLASTN searches (Altschul et al. 1997). An arbitrary expect score cutoff of <e −05 was used to classify AtCXE genes. Additional carboxylesterases from the Oryza sativa, Saccharomyces cerevisiae, and Synechocystis sp. PCC 6803 genomes were searched for using sequences deposited in GenBank. The search for AtCXE genes was conducted during March 2002, while the search for O. sativa, S. cerevisiae, and Synechocystis carboxylesterase genes was conducted in May 2002.
A multiple sequence alignment of the predicted AtCXE peptides was created with CLUSTALX (v1.81) using the default settings (Thompson et al. 1997). The following 33 GenBank protein sequence accessions were also included in the CLUSTALX alignment: Arabidopsis thaliana (see Table 1 for the 20 AtCXE accessions); Capsicum annuum (AAF77578.1, PepEST); Lycopersicon esculentum (BAA74434.1, Lehsr203J); Nicotiana tabacum (AAF62404.1, hsr203J): Oryza sativa (BAB44070.1. OsCXEl; BAB44069.1, OsCXE2; CAD40857.1, OsCXE3; BAB44059.1, OsCXE4; BAB90534.1, OsCXE5): Pinus radiata (AAD04946.2, PrMC3): Pisum sativum (BAA85654.1. E86); Saccharomyces cerevisiae (NP_010716.1, ScCXE1; NP_011779.1, ScCXE2); and Synechocystis sp. PCC 6803 (NP_440291.1, SynCXE1). Shading of the conserved amino acids was conducted using GeneDoc (v2.6.002 [Nicholas and Nicholas 1997]).
Phylogenetic analysis was carried out using the PHYLIP software package (Felsenstein 1993). Terminal gaps in the peptide alignment produced using CLUSTALX (above) were removed prior to running the analysis, while the internal alignment gaps were left and analysis conducted scoring gaps as characters or as missing characters. Distances were calculated using the Dayhoff matrix (PROTDIST), and the neighbor joining method was used to create the tree (NEIGHBOR). Parsimony trees were constructed using PROTPARS implemented in PHYLIP. Bootstrap analysis was conducted using 1000 bootstrap replicates (Felsenstein 1993). TreeView (v.1.6.6) was used to display resulting trees (Page 1996).
GenomePixelizer (January 7, 2002) was used to create a genomic map of the AtCXE gene family (Kozik et al. 2002). The coordinates for the AtCXE family and the Arabidopsis matrix file were obtained from information included with the program. Relationships among the AtCXE family members were analyzed at a 40% and greater amino acid identity threshold.
Gene Expression Characterization
Tissue for RT-PCR analysis was collected from Arabidopsis ecotype Columbia. Plants were grown in soil under glasshouse conditions at 18–25°C under 14 h of daylight. Genomic DNA was extracted from 200 mg of rosette leaves (˜4 weeks old) using the protocol described by Li and Chory (1998). RNA was extracted from various tissues (young root— roots from six-true leaf-stage plants; mature root—roots from bolted plants; two-, four-, and six-leaf seedlings—aerial parts from two-, four-, or six-true leaf-stage plants; leaf—rosette leaves from bolted plants; stem—stem from bolted plants with petioles and floral structures removed; flower bud—inflorescence apex; flower; young siliques—flat green siliques; mature siliques—green siliques filled with seed) and pooled from ˜10 plants using the following protocol. Tissue (200–500 mg) was ground under liquid N2. Phenol (250 µl) plus 250 µl of RNA extraction buffer (100 mM LiCl, 100 mM Tris–HCl, pH 8, 10 mM EDTA, pH 8, and 1% SDS [w/v] final concentration) was preheated to 80°C before being mixed for 30 s with ground tissue. Chloroform (250 µl) was added to the tissue extraction solution, vortexed, and then spun at 20,000g for 5 min at 4°C. RNA was precipitated from the supernatant with 1 vol of 4 M LiCl overnight on ice before being spun at 20,000g for 10 min at 4°C. The pellet was resuspended in 500 µl of 2 M LiCl for 30 min on ice and then spun at 20,000g for 5 min at 4°C. The RNA pellet was then resuspended in 100 µl of 1% L-Sarcosyl and extracted once with phenol:chloroform:isoamyl alcohol (25:24:1). RNA in the supernatant was precipitated with 0.1 vol of 3 M sodium acetate, pH 4.8, and 2.5 vol of 95% ethanol, washed in 70% ethanol, dried, and then resuspend in 20 µl of DEPC-treated water.
Contaminating genomic DNA was removed from total RNA by digestion with DNasel (Invitrogen) following the manufacturer’s protocol. First-strand cDNA was synthesized from 1 µg of total RNA primed with 100 µmol RoR1dT16 (5′-ATCGATGGTCGACGCATGCGGATCCAAAGCTTGAATTCGAGCTCT15-3′) and 250 ng random hexamers (Promega) following manufacturer’s instructions for Superscript II Reverse Transcriptase (Invitrogen). A negative control for each tissue analyzed was performed as above, with water used in place of the reverse transcriptase enzyme.
Gene specific PCR primers were designed to encompass most of the predicted coding region for each AtCXE gene (see Table 2 for details). PCR experiments were performed a minimum of two times on RNA preparations from two independent tissue samples. PCR amplification reactions were performed in 25-µl volumes containing 0.2 mM dNTPs, 0.6 µM of each primer pair, 1.5 mM MgCl2, 1× Taq buffer (Invitrogen), and 1 unit of Taq DNA polymerase (Invitrogen), plus 2 µl of genomic DNA (˜10 ng) or cDNA (equivalent to 10 ng of total RNA) as template. The PCR cycling program had an initial denaturation step of 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 3 min. The program ended with a final extension cycle of 72°C for 5 min. Control PCR reactions with the control “cDNA” templates (i.e., no reverse transcriptase added during cDNA synthesis) were performed as described above. Resulting products were resolved on 1.2% agarose gels. Predicted PCR product sizes for each gene are listed in Table 2.
The identity of a selection of amplified gene products, generated using the AtCXE primer pairs above, was verified by DNA sequencing. Genomic DNA was amplified for eight AtCXE genes (AtCXE2, 10, 11, 14, 15, 16, 19, and 20) using the primers described above. PCR products from these reactions were purified using QIAquick PCR Purification Columns (Qiagen). DNA sequencing reactions were carried out using Dynamic ET Sequencing Dye (Amersham Biosciences) according to manufacturer’s instructions. Primers used to sequence purified PCR products were as follows (primers are written in a 5′-to-3′ direction, primer names correspond to the AGI gene code presented in Table 1): At1g47480.2f TTGTCAATCAGGCTAACGTC, At3g05120.2f CTTGTGCTT ATGATGATGGT, At3g27320.2f AGGTGAGGTGAAGAAAT CAG, At3g63010.2f GTCAACCTTAACGAATGCAA, At5g06570.2f CTGGTTTGAAGATGGAACAG, At5g14310.2f TGT TCAAGGACAGATTGTGG, At5g27320.2f GTTAATCTTATT GAGAGCAAG, and At5g62180.2f ATCTCCTTCGTACCGAT TAG.
Results and Discussion
Identification of the AtCXE Gene Family in Arabidopsis
The nomenclature for carboxylesterases has been a source of detailed discussion (e.g., Eeiner et al. 1989). Most previous reports have used functional criteria to classify the carboxylesterases. In the absence of any biochemical or physiological data, we have defined the Arabidopsis carboxylesterase family as a group of phylogenetically related sequences and use the abbreviation AtCXE.
Mining the complete Arabidopsis genome (The Arabidopsis Genome Initiative 2000) reveals 20 carboxylesterase genes (AtCXE1–20). A summary of the features and identifiers for the AtCXE gene family is provided in Table 1. Numbering of the AtCXE family is based on the ordering of the protein identifier code assigned by The Arabidopsis Genome Initiative (AGI [see Schoof et al. 2002]). The AtCXE family has an average amino acid identity of 34.1% and ranges from 22.8 to 86.0% identity.
The 20 AtCXE genes were identified through a combination of key word searches and manual iterative TBLASTN searches, starting with the previously published plant carboxylesterases. The Carbohydrate-Active Enzymes web page (CAZy; http://afmb.cnrs-mrs.fr/~cazy/CAZY/index.html ) contains a list of esterases from a variety of organisms, including Arabidopsis. This on-line resource is dedicated to describing “the families of structurally-related catalytic and carbohydrate-binding modules (or functional domains) of enzymes that degrade, modify, or create glycosidic bonds.” The list of AtCXE genes found from our BLAST search was similar to the list in CAZy (family 10). Four additional Arabidopsis genes were predicted to be carboxylesterases in CAZy, which have not been included in our analysis (AGI protein code: At1g26120, At3g02410, At5g15860, and At5g36210). Three of these genes (At1g26120, At3g02410, At5g15860) are closely related to each other and contain a catalytic triad. However, they are divergent from the 20 AtCXE genes and other plant carboxylesterases and lack sequence conservation surrounding the motif known as the oxyanion hole (contains a TGG motif rather than the HGG motif seen for AtCXE and the other plant carboxylesterases). Interestingly, these three genes (At1g26120, At3g02410, and At5g15860) possess a large number of introns (10, 8, and 9 introns, respectively) with many shared intron positions. The genes At1g26120, At3g02410, and At5g15860 are postulated to be lipases based on the automated annotation within the MIPS Arabidopsis thaliana Database (MatDB [Schoof et al. 2002]). The fourth gene, At5g63210, is likely to be a carboxypeptidase based on BLASTP analysis. It contains a unique exon/ intron structure (18 introns) and a significantly longer predicted peptide sequence (678 amino acids).
Annotation of the exon/intron boundaries for the 20 AtCXE genes from MatDB was compared with available expressed sequence tag (EST) information (see Table 1). MatDB annotations were correct in all but one instance. Exon 2 of AtCXE16 (At5g14310) begins at base 1791 rather than base 1812. The new cDNA sequence includes ATGTGTTCTTCTCG GGGTGAG at the 5′ end of exon 2. This correction is based on EST information from GenBank Accessions AV550947 and H76498 (original clone names are RZ119f03R and 196M16T7, respectively).
Based on alignment with available EST information, 13 of the 20 AtCXE genes have no introns. The remaining seven AtCXE genes contain a single intron (Table 1). One intron site is shared by AtCXE10, 14, and 19 and is located between bases 39/40 of the predicted cDNA sequence (A of the translation start codon ATG = 1). A second intron site is shared by AtCXE11 and 16. The intron is located between bases 845/846 for AtCXE11 and 800/801 for AtCXE16. The precise cDNA location is not conserved due to insertion/deletion events within AtCXE11 and 16. Alignment of the translated cDNA sequences for AtCXE11 and 16 reveals a high degree of amino acid conservation surrounding the intron splice site: 18 of 20 amino acids are conserved upstream from the “intron splice site” position and 19 of 20 amino acids downstream of the “intron splice site” position are conserved. The remaining two genes (AtCXE2 and AtCXE15) possess unique intron sites. Intron positions for AtCXE2 and 15 are located between bases 351/352 and 683/684, respectively.
Sequence Alignment of the AtCXE Family
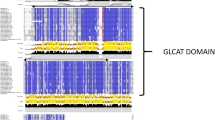
An alignment of the predicted amino acid sequences for the 20 AtCXE genes is presented in Fig. 1. The alignment also includes 10 other plant carboxylesterases (full length) found in GenBank as of May 2002. The sequence alignment reveals amino acid motifs and secondary structural features characteristic in members of the α/β hydrolase fold superfamily and more particularly of carboxylesterases (Fig. 1).

Multiple amino acid sequence alignment of plant carboxylesterases. Abbreviations for species and carboxylesterase names are as follows: AtCXE1 to 20 (Arabidopsis thaliana carboxylesterase; see Table 1 for AtCXE protein accessions); CaPepEST (Capsicum annuum PepEST; AAF77578.1); Nthsr203J (Nicotiana tabacum hsr203J; AAF62404.1); Lehsr203J (Lycopersicon esculentum hsr203J; BAA74434.1); OsCXE1 to 5 (Oryza sativa carboxylesterase; BAB44070.1, BAB44069.1, CAD40857.1, BAB44059.1, and BAB90534.1); PrMC3 (Pinus radiata PrMC3; AAD04946.2); PsE86 (Pisum sativum E86; BAA85654.1); ScCXE1 and 2 (Saccharomyces cerevisiae carboxylesterase; NP_010716.1 and NP_011779.1); SynCXE1 (Synechocystis sp. PCC 6803 carboxylesterase; NP_440291.1). Conserved motifs are marked above the alignment. O—oxyanion hole; S—catalytic serine; D—active-site acidic residue (primarily aspartate, but glutamate is also present); H—active—site histidine. Predicted β-strands (arrows β1–8) and α-helices (cylinders α1–5) are numbered. Shading of amino acids indicates degree of conservation: black—100% amino acid conservation: dark gray—70–99% amino acid conservation. Amino acid position for individual genes is marked on the right, while the consensus alignment position is indicated along the top.
Carboxylesterases contain a catalytic triad that is made up of a nucleophilic serine, an acidic residue (aspartate or glutamate), and a histidine (Ollis et al. 1992). These active-site residues are found dispersed within the primary peptide sequence but are positioned adjacent to one another in the tertiary structure (Ollis et al. 1992). Within the basic core structure of the α/β hydrolase fold, the catalytic serine is located in the loop immediately after β-strand 5, the acidic residue (i.e., aspartate or glutamate) follows β-strand 7, and the histidine trails the final β-strand (Ollis et al. 1992). Juxtaposition of all three active-site residues within the folded protein is necessary for activation of the catalytic serine. Activation of the serine occurs through a charge relay system where the acidic residue draws a proton from the histidine, which in turn draws a proton from serine to create a nucleophilic serine (Oilis et al. 1992).
The plant carboxylesterases analyzed here are predicted to share the core α/β hydrolase fold positions for the catalytic triad (i.e., serine, acidic residue, and histidine), as shown in Fig. 1. The predicted serine and acidic residues are both highly conserved in our alignment. The acidic residue for most of the plant carboxylesterases is an aspartate. All but one AtCXE contain an aspartate in the active site. The active-site acid residue for AtCXE15 is a glutamate. In contrast, the acidic residue in animal kingdom carboxylesterases is typically a glutamate (Cygler et al. 1993).
The position of the histidine is not completely conserved in plants with regard to the aligned amino acid sequences. The predicted active site histidine of AtCXE10, 14, and 19 is found further C-terminal in the peptide alignment (Fig. 1). Manipulation of the multiple alignment parameters in CLUSTALX (v1.81) did not improve the alignment. It remains to be determined if these enzymes (AtCXE10, 14, and 19) have carboxylesterase activity. An active-site histidine cannot be found for OsCXE2 within the expected region (Fig. 1). Since a histidine forms an essential portion of the catalytic site, OsCXE2 may not possess carboxylesterase activity. Another possibility for the apparent loss of the active-site histidine is that the primary sequence order for the OsCXE2 catalytic triad residues may differ from the model, and therefore the OsCXE2 histidine would be located elsewhere in the primary amino acid sequence. Precedence for variations in the primary sequence order for the catalytic triad residues within the α/β hydrolase superfamily can be found for trypsin, papain, and subtilisin (Ollis et al. 1992). Rearrangement of the catalytic triad order would, however, be highly unusual for a carboxylesterase.
The catalytic serine residue for members of the α/β hydrolase fold superfamily is surrounded by the characteristic pentapeptide motif G–X–S–X–G. This motif extends the serine into the center of the active site through a tight bend directly after the fifth β-strand and is often referred to as the nucleophilic elbow (Ollis et al. 1992). The G–X–S–X–G pentapeptide motif is completely conserved within the carboxylesterases from Arabidopsis and the other plant carboxylesterases (Fig. 1). Serine is an interesting amino acid because it is encoded by six codons split into two codon groups, AGY (where Y = T or C) and TCN (N = A, T, C, or G). Two mutations are required to switch from one group to the other. Therefore, unless both mutations occur simultaneously, the intermediate protein does not contain a serine at this position and is likely to be nonfunctional. Examination of the codon usage of the active-site serine for the AtCXE genes reveals a nearly exclusive use of the AGY serine. AtCXE15 is the only exception to this, using the TCN serine codon group. Animal esterases predominantly use the TCN serine codon group (Petersen and Drablos 1996).
The oxyanion hole is another important structural feature of the carboxylesterase family. This structure is involved in stabilizing the substrate–enzyme tetrahedral intermediate during hydrolysis through interactions between the oxyanion produced in the intermediate with three backbone nitrogen atoms (Ollis et al. 1992). A portion of the oxyanion hole is made up of nitrogens from two adjacent glycine residues located between β-strand 3 and α-helix 1 contained within a conserved HGG box motif shared across all but one of the plant carboxylesterase genes examined. The fourth residue after each HGG is either a glycine, a serine, or an alanine (Fig. 1). A notable exception to the conservation of the HGG motif is found in AtCXE9, which has a HSG motif. It is interesting to speculate whether this change (i.e., substitution of an aliphatic amino acid for a hydroxylated residue) affects the catalytic ability of this putative carboxylesterase. The remaining backbone nitrogen involved in the oxyanion interaction comes from the residue following the active-site serine and is often an alanine in the plant carboxylesterases (Fig. 1).
Phylogenetic Analysis of the AtCXE Gene Family
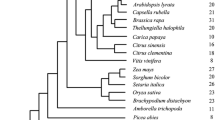
Phylogenetic analysis of the plant carboxylesterases has been conducted using the alignment presented in Fig. 1 (see Fig. 2). This distance-based tree revealed seven clades, numbered arbitrarily in an anticlockwise manner. The groupings are well supported by bootstrap analysis, with five showing bootstrap values of 95% or greater. The AtCXE genes are represented in six of the seven groups (clades I–VI). The five OsCXE genes are found in three clades (I, II, and V). One clade (VII) does not contain any Arabidopsis or O. sativa genes. The single Synechocystis and two S. cerevisiae carboxylesterases have been used as an outgroup for this analysis. The assignment of the carboxylesterase genes to the seven groups from trees built using data obtained from parsimony analysis was the same as for distance analysis (data not shown).

Unrooted neighbor- joining tree showing the phylogenetic relationship of the plant carboxylesterases. Branch nodes showing 50% or greater bootstrap support are labeled (percentage of 1000 bootstrap replicates). The seven plant carboxylesterase clades are shaded gray and labeled with roman numerals. Species and carboxylesterase name abbreviations are presented in the legendto Fig. 1.
The largest group is clade III, possessing 40% (8 of 20) of the AtCXE genes (AtCXE1, 2, 3, 4, 5, 7, 12, 13) and OsCXE3. Given the degree of sequence conservation and short branch lengths, it seems likely that subgroups within clade III have recently undergone an expansion/duplication (e.g., AtCXE3, 4, 5 and AtCXE1, 7, 12, 13).
Three clades (II, IV, and V) contain four taxa each. Clade II contains three Arabidopsis genes (AtCXE8, 9, and 20) and a carboxylesterase from C. annuum (PepEST) that is involved in plant–pathogen interactions. Clade IV consists entirely of AtCXE genes (10, 14, 18, and 19). Clade V shows an interesting grouping with three of the five O. sativa genes (OsCXE1, 2, and 4) and AtCXE15. An O. sativa gene is also found in each of clade I (OsCXE5) and clade III (OsCXE3). Given that the publicly available O. sativa genome sequence is not yet complete, further O. sativa genes may be found that associate with the other clades.
Three plant taxa have representative carboxylesterases in each of clades I, VI, and VII. AtCXE11 and 16 plus OsCXE5 make up clade I. An insertion of approximately 60 amino acids located between β-strands 2 and 3 and a second insertion (˜30 amino acids) positioned between α-helix 2 and β-strand 5 is characteristic for the AtCXE genes in this clade (refer to Fig. 1). Clade VI consists of two genes from Arabidopsis (AtCXE6 and 17) and a single gene from P. radiata (PrMC3). PrMC3 (from clade VI) was identified in a screen for genes specifically expressed in the male cone; however, no functional data are available for PrMC3 (Walden et al. 1999). Further characterization of PrMC3 is required to test an involvement with male reproduction. Clade VII is made up of three highly similar genes from N. tabacum (Nthsr203J), L. esculentum (Lehsr203J), and P. sativum (E86). There are no AtCXE genes present in this clade. All of the genes in clade VII have been associated with plant–pathogen interactions (Pontier et al. 1994, 1998; Ichinose et al. 2001). The split between clades VII and VI has bootstrap support of only 52%. These clades may in fact represent a single grouping. The deep branching pattern suggests that these clades (VI and VII) either diverged a long time ago or have elevated rates of evolution. Another gene associated with plant–pathogen interactions is PepEST (clade II) from C. annuum, which is phylogenetically distinct from the hsr203J genes from N. tabacum, L. esculentum, and P. sativum (clade VII). The clade II Arabidopsis genes (AtCXE8, 9, and 20) are closely related to PepEST, suggesting that they may also be involved in plant–pathogen interactions (see later for further discussion).
From the phylogenetic analysis, predictions can be made concerning which AtCXE genes duplicated to expand the six clades with Arabidopsis representatives. For example, duplications of an AtCXE2-like ancestor have given rise to two distinct subgroups within clade III, with one subgroup containing AtCXE3, 4, and 5 and the second containing AtCXE1, 7, 12, and 13. In clade IV, an AtCXE18-like ancestor appears to have undergone duplications to give rise to AtCXE10, 14, 19.
Genomic Arrangement of the AtCXE Gene Family
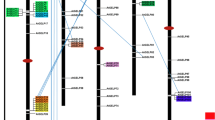
The software program GenomePixelizer was used to map the AtCXE gene family and their relationships onto the five Arabidopsis chromosomes (Kozik et al. 2002). This analysis allows classification of the duplication events into tandem, intrachromosomal, or interchromosomal events. The structure of the predicted gene duplications within the AtCXE family is illustrated in Fig. 3. The 20 AtCXE genes are distributed across chromosomes I, II, III, and V, but are absent from chromosome IV. From Fig. 3, the AtCXE genes cluster into approximately five broad groups, plus two orphan genes (AtCXE15 and 18). The groups presented in Fig. 3 are as follows: AtCXE11 and 16 (clade I); AtCXE8, 9, and 20 (clade II); and AtCXE1, 2, 3, 4, 5, 7, 12, and 13 (clade III); AtCXE10, 14, and 19 (clade lV); and AtCXE6 and 17 (clade VI). Additionally, tandem duplication events have occurred for AtCXE3, 4, and 5 (clade III); AtCXE12 and 13 (clade III); and AtCXE8 and 9 (clade II). Only AtCXE18 (clade IV) did not follow the grouping as determined from the phylogenetic analysis (see Fig. 2), being represented as an orphan gene in the GenomePixelizer analysis (Fig. 3).

Genomic organization of the AtCXE genes as analyzed using GenomePixelizer (Kozik et al. 2002). Thin black lines join genes that have a ≥40% predicted amino acid sequence identity. Related AtCXE genes that show ≥50% predicted amino acid sequence identity are as follows: 7 with 3, 4, and 5 (50–60%); 1 with 7 (60–70%); 10 with 14 (60–70%); 14 with 19 (70–80%); 11 with 16 (70–80%); 12 with 13 (70–80%); 3 with 4 (70–80%); 3 with 5 (70–80%); 4 with 5 (70–80%); and 10 with 19 (80–90%). AtCXE members belonging to the same gene cluster are outlined by the same shape. The five Arabidopsis chromosomes are labeled I–V, with the AtCXE genes represented as small black boxes along the chromosomes. Arrows denote gene orientation along the chromosome.
In the AtCXE family only three tandem duplications can be identified. These include 7 of the 20 genes: AtCXE3, 4, and 5 (clade III); AtCXE8 and 9 (clade II); and AtCXE12 and 13 (clade III). The orientation of the genes within each tandemly duplicated cluster is maintained. Clusters of tandemly duplicated genes have been observed for other gene families within the Arabidopsis genome (e.g., Richly et al. 2002). However, the majority of predicted duplication events of AtCXE genes are not tandem, either occurring within (over a large distance) or between chromosomes. In other organisms many of the duplication events for carboxylesterases are tandem. For example, in Drosophila most of the genes are arranged in clusters (Oakeshott et al. 1999). The D. melanogatser α-esterase cluster is made up of 10 active esterases plus one pseudogene spanning a 60-kb chromosomal region (Russell et al. 1996). Tandem duplication has also been identified as the manner of expansion for the β-esterase cluster, which contains two genes in D. melanogaster and three in related taxa (Oakeshott et al. 1995). Similarly for Caenorhabditis elegans, 29 of the 49 carboxylesterases map to just six chromosomal loci (Oakeshott et al. 1999). In the mosquito, Anopheles gambiae, clustering of esterases is even more pronounced. Only 16 of the 51 carboxylesterase genes are found as singletons, the rest being represented in clusters, with one cluster containing 23 carboxylesterase genes (Ranson et al. 2002). The pattern of nontandem duplication found in Arabidopsis implies one of two possibilities: that the duplications themselves were interchromosomal in the first instance or that the original duplications events were tandem but over time the tandem arrays have been broken up by rounds of genome duplication with subsequent loss and rearrangement. While the details of the genome duplication history for Arabidopsis are still under debate, it is clear that Arabidopsis has undergone more than one round of whole-genome duplication, with many smaller-scale translocation events occurring over its history (Ku et al. 2000; Blanc et al. 2000; Vision et al. 2000; The Arabidopsis Genome Initiative 2000; Simillion et al. 2002).
To gain insight into how the AtCXE family may have expanded, the AtCXE gene positions were mapped to published duplication events within the Arabidopsis genome (The Arabidopsis Genome Initiative 2000). Approximately 60% of the Arabidopsis genome is found within 24 large duplications, with 17% of the genes found in tandem arrays (The Arabidopsis Genome Initiative 2000). Chromosomal duplications within the AtCXE gene family do not correspond to the known major chromosomal duplications across the Arabidopsis genome reported by The Arabidopsis Genome Initiative (2000; data not shown). Although the AtCXE genes do not map to previously reported Arabidopsis chromosomal duplication sites, it is possible that smaller chromosomal duplication events have occurred that were not detected in these previous reports. Inspection of 10 genes on either side of each AtCXE locus was performed to identify potential small chromosomal duplication events. The genes flanking the AtCXE loci were compared at a 30% amino acid identity threshold. Two small duplications were identified that included AtCXE genes. A conserved block of three genes is shared around AtCXE11 (on chromosome III) and AtCXE16 (on chromosome V). The shared pairs of genes are At3g27255 and At5g14270, At3g27270 and At5g14280, and At3g27280 and At5g14300 on chromosomes III and V, respectively. A second small duplicated region was also observed around AtCXE10 and 19, where an ordered group of genes is shared. These shared gene pairs are as follows: At3g05230 and At5g27430; At3g05200 and At5g27420; At3g05190 and At5g27410; At3g05150 and At5g27360; and At3g05150 and At5g27350, At5g27360. Genes At5g27350 and At5g27360 appear to be the product of an additional round of duplication. AtCXE14 is grouped in clade IV with AtCXE10 and 9. However, the region surrounding the AtCXE14 locus does not share any of the conserved surrounding genes found for the AtCXE10 and 19 loci. Together these observations suggest that two small independent chromosomal translocation events have occurred between chromosomes III and V that were not detected in the analysis performed by The Arabidopsis Genome Initiative (2000).
AtCXE Gene Expression Patterns
RT-PCR was used to investigate expression patterns for the AtCXE genes over a range of tissues and developmental stages from Arabidopsis using PCR primers designed to each individual AtCXE gene. All amplified products were of a size predicted from the AtCXE genes, and the DNA sequence from eight chosen amplified genomic DNA products matched with the expected AtCXE genes. The PCR conditions used in this experiment were designed to detect the presence or absence of expression and not for quantitative interpretations. However, faintly visible bands are most likely due to genes being expressed at a lower level than the more intense bands.
All 20 AtCXE genes are transcribed (Fig. 4). Control reactions lacking reverse transcriptase were conducted to demonstrate the absence of any contaminating genomic DNA for all of the original RNA samples. Amplified products were never observed in any of these control experiments. All tissue samples tested were positive for expression of each AtCXE gene, except where specifically mentioned. Samples showing either trace amounts or no product were confirmed by increasing the number of cycles from 30 to 40 (data not shown). Thirteen of the 20 AtCXE genes are detected across all tissue samples analyzed: AtCXE11 and 16 (clade I); AtCXE20 (clade II); AtCXE2, 4, 5, 6, and 12 (clade III); AtCXE10, 14, 18, and 19 (clade IV); AtCXE15 (clade V); and AtCXE6 (cladeVI). The following genes are not detected in roots, being observed only in aerial portions of the plant: AtCXE8 and 9 (clade II); AtCXE3, 7, and 13 (clade III); and AtCXE17 (clade VI). The most striking expression pattern is observed for AtCXE3 (clade III) and AtCXE9 (clade II). AtCXE3 and 9 are only detected in flowers and siliques; however, AtCXE9 was never observed in mature siliques, even after 40 cycles of amplification (data not shown). The only other genes not detected in all tissues were AtCXE1 and AtCXE13 (both from clade III). AtCXE1 is absent from the aerial parts of seedlings and from leaves on bolted plants, while AtCXE13 is not expressed in leaf or stem tissue from bolted plants. The overlapping pattern of expression shown by the AtCXE genes suggests that the activity for some proteins may be functionally redundant. However, certain AtCXE genes with overlapping expression patterns may be expressed in different cell types within each tissue and possess different biological functions.

Expression pattern of the AtCXE family examined by RT-PCR. The tissues from which RNA was derived are presented along the top of the diagram. Control reactions were as follows: water, water template as a negative control for PCR; and gDNA, genomic DNA template as a positive control for PCR. The third control reaction used primers specific to actin (ACT2), as this gene is constitutively expressed across all tissues and developmental stages (bottom of figure). Amplified product sizes correspond to those predicted from the gene sequences (see Table 2). The tree to the left of the expression data is the same as from Fig. 2 but is represented here as a rectangular cladogram drawn using ScCXE1, ScCXE2, and SynCXE1 as the outgroup. Gray shading and roman numerals correspond to plant carboxylesterase clades identified in Fig. 2. Species and carboxylesterase name abbreviations are presented inthe figure legend to Fig. 1.
Combining phylogenetic and expression pattern data has been a successful approach toward gaining insight into the potential function of genes within a gene family (e.g., MYB gene family [Stracke et al. 2001]). The expression data for the AtCXE genes have been mapped onto the tree from Fig. 2 to produce Fig. 4. However, the broad overlapping patterns of expression for the AtCXE genes do not correlate with groups identified in the phylogenetic analysis (Fig. 4). For example in clade II, AtCXE20 is expressed across all tissues, AtCXE8 is not expressed in the roots, and AtCXE9 is only expressed in the flowers and siliques. In another example, genes within clade III show wide variation in expression patterns: AtCXE2, 4, and 5 are detected in all tissue samples; AtCXE7 and 13 are not expressed in the roots; and AtCXE3 is only found in flowers and siliques. It seems that flower/silique expression has evolved twice within the AtCXE family, once in AtCXE3 (clade III) and again in AtCXE9 (clade II). Comparison of more fine-scale expression patterns among the AtCXE genes will require more precise dissection of tissues or reporter studies.
Recent reports suggest a correlation between the physical linkage of genes and their coexpression patterns (for examples see Boutanaev et al. 2002; Caron et al. 2001; Cohen et al. 2000; Roy et al. 2002; Spellman and Rubin 2002). An examination of the closely linked AtCXE family members (i.e., AtCXE3, 4, and 5; AtCXE8 and 9; and AtCXE12 and 13) and their corresponding expression patterns does not provide evidence for coregulation of these clustered AtCXE genes. For example, in clade II AtCXE9 is expressed only in the flowers and siliques, whereas AtCXE8 is detected in the aerial portions of the plant (see Figs. 3 and 4).
Speculations on Function
Four of the plant carboxylesterases studied to date have been implicated in plant–pathogen interactions. Clade VII is entirely made up of hsr203J orthologs (Fig. 3). The N. tabacum and L. esculentum hsr203J orthologs are specifically expressed during the hypersensitive response (HR) after exposure to plant pathogens (Pontier et al. 1994, 1998). Experiments with the N. tabacum hsr203J and L. esculentum hsr203J genes have shown that hsr203J expression is tightly associated with tissues undergoing programmed cell death. Both HR (mediated by resistance gene recognition of avirulence proteins) and apoptosis (induced by heavy metals) resulted in increased expression of hsr203J. Expression was not altered by exposure to signaling molecules that are induced as a downstream consequence of HR (e.g., H2O2, salicylic acid [Pontier et al. 1998]). While it is true that AtCXE genes are not represented in clade VII, a role associated with programmed cell death is still possible for AtCXE6 or 17 if clades VI and VII are the same group.
PepEST is a carboxylesterase gene that is also upregulated during plant–pathogen interactions, however, it is found in a separate clade (clade II) from the hrs203J orthologs (Kim et al. 2001). AtCXE8, 9, and 20 are also members of clade II and may also be involved in plant–pathogen interactions. The mechanism for PepEST is not yet known, but given the parallels between PepEST and the hsr203J genes (i.e., upregulated during plant–pathogen interactions), it is not unreasonable to suggest a similar role for PepEST. The functional role for PrMC3 is unknown. However, based on similarity to hsr203J and its presence in the developing male cone, the authors speculated a role for PrMC3 in programmed cell death during developmental cell turnover (Walden et al. 1999). Our phylogenetic analysis supports its close relationship to the hsr203J homologs and together with AtCXE6 and 17 support a hypothesis for involvement of these genes in cell death.
Many other biological functions are possible for members of the AtCXE family particularly based on precedents from other systems. Hydrolysis of xenobiotics is an important role that carboxylesterases perform in other organisms (e.g., Satoh and Hosokawa 1998). In other systems, carboxylesterases involved in detoxification are often constitutively expressed (Sandermann 1992). The constitutive expression of many of the AtCXE genes is consistent with such a role. For this function, expression in at least the roots would also be expected.
Carboxylesterases are also known to be involved in signaling pathways, often resetting the system by hydrolyzing the signalling molecule: for example, sex pheromones in moths (Vogt et al. 1985) and the neural transmitter acetylcholine in animals (Taylor and Radic 1994). Carboxylesterases may also have a role to play in plant signaling pathways. An esterase from L. esculentum has recently been identified that is able to cleave the signaling molecule methyl jasmonate (Stuhlfelder et al. 2002). While the partial amino acid sequence for the methyl jasmonate cleaving enzyme suggests that it is not related to the AtCXE gene family, a role in plant signaling pathways for carboxylesterases seems possible. Other possible substrates for plant carboxylesterases include esters produced by plants to attract pollinators/seed dispersers and to deter herbivores (e.g., hexanoate, Z-3-hexenyl acetate [Pichersky and Gershenzon 2002]).
While we have identified some possible roles for Arabidopsis carboxylesterases, extensive biochemical and physiological studies will be needed to determine the exact functions they perform in plants. Arabidopsis, with its extensive repertoire of available genetic resources, will be well suited to determine these roles.
References
SF Altschul TL Madden AA Schäffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25 3389–3402 Occurrence Handle9254694
G Blanc A Barakat R Guyot R Cooke M Delseny (2000) ArticleTitleExtensive duplication and reshuffling in the Arabidopsis genome. Plant Cell 12 1093–1101 Occurrence Handle1:CAS:528:DC%2BD3cXlslygtL0%3D Occurrence Handle10899976
R Boskovic KR Tobutt (1998) ArticleTitleInheritance and linkage relationships of isoenzymes in two interspecific cherry progenies. Euphytica 103 273–286 Occurrence Handle10.1023/A:1018664600714 Occurrence Handle1:CAS:528:DyaK1cXntlSqsr0%3D
AM Boutanaev AI Kalmykova YY Shevelyov DI Nurminsky (2002) ArticleTitleLarge clusters of co-expressed genes in the Drosophila genome. Nature 420 666–669 Occurrence Handle10.1038/nature01216 Occurrence Handle1:CAS:528:DC%2BD38XpsVSitbk%3D Occurrence Handle12478293
H Caron B van Schaik M van der Mee F Baas G Riggins P van Sluis MC Hermus R van Asperen K Boon PA Voute S Heisterkamp A van Kampen R Versteeg (2001) ArticleTitleThe human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science 291 1289–1292 Occurrence Handle1:CAS:528:DC%2BD3MXhtlSgs70%3D Occurrence Handle11181992
BA Cohen RD Mitra JD Hughes GM Church (2000) ArticleTitleA computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet 26 183–186 Occurrence Handle10.1038/79896 Occurrence Handle1:CAS:528:DC%2BD3cXntlGms70%3D Occurrence Handle11017073
M Cygler JD Schrag JL Sussman M Harel I Silman MK Gentry BP Doctor (1993) ArticleTitleRelationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci 2 366–382 Occurrence Handle1:CAS:528:DyaK3sXksVCitrg%3D Occurrence Handle8453375
A Dansi HD Mignouna J Zoundjihekpon A Sangare R Asiedu N Ahoussou (2000) ArticleTitleUsing isozyme polymorphism to assess genetic variation within cultivated yams (Dioscorea cayenensis/Dioscorea rotundata complex) of the Republic of Benin. Genet Resour Crop Evol 47 371–383 Occurrence Handle10.1023/A:1008718411962
E Eeiner WN Aldridge FCG Hoskin (1989) Enzymes hydrolysing organophosphorous compounds. John Wiley & Sons New York
JC Fachinello S Musacchi S Zuccherelli S Sansavini (2000) ArticleTitleIsoenzymatic variability in pear tissues for fingerprinting. Pesqui Agropecu Bras 35 1427–1432
Felsenstein J (1993) PHYLIP—Phylogeny inference package (version 3.6a2). Distributed by the author (evolution.genetics. washington.edu/phylip.html)
J Hemingway (2000) ArticleTitleThe molecular basis of two contrasting metabolic mechanisms of insecticide resistance. Insect Biochem Mol Biol 30 1009–1015 Occurrence Handle10.1016/S0965-1748(00)00079-5 Occurrence Handle1:CAS:528:DC%2BD3cXmtlOgtr8%3D Occurrence Handle10989287
E Huala A Dickerman M Garcia-Hernandez D Weems L Reiser F LaFond D Hanley D Kiphart J Zhuang W Huang L Mueller D Bhattacharyya D Bhaya B Sobral B Beavis C Somerville S Rhee (2001) ArticleTitleThe Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res 29 102–105 Occurrence Handle1:CAS:528:DC%2BD3MXjtlWntLo%3D Occurrence Handle11125061
Y Ichinose Y Hisayasu S Sanematsu Y Ishiga H Seki K Toyoda T Shiraishi T Yamada (2001) ArticleTitleMolecular cloning and functional analysis of pea cDNA E86 encoding homologous protein to hypersensitivity-related hsr203J. Plant Sci 160 997–1006 Occurrence Handle10.1016/S0168-9452(01)00343-0 Occurrence Handle1:CAS:528:DC%2BD3MXisF2kurc%3D Occurrence Handle11297797
AL Kahler RW Allard (1970) ArticleTitleGenetics of isozyme variants in barley. I. Esterases. Crop Sci 10 444–448 Occurrence Handle1:CAS:528:DyaE3cXlt1Ort7s%3D
YS Kim HH Lee MK Ko CE Song CY Bae YH Lee BJ Oh (2001) ArticleTitleInhibition of fungal appressorium formation by pepper (Capsicum annuum) esterase. Mol Plant Microbe Interact 14 80–85 Occurrence Handle1:CAS:528:DC%2BD3MXot1Oh Occurrence Handle11194875
A Kozik E Kochetkova R Michelmore (2002) ArticleTitleGenomePixelizer—A visualization program for comparative genomics within and between species. Bioinformatics 18 335–336 Occurrence Handle10.1093/bioinformatics/18.2.335 Occurrence Handle1:CAS:528:DC%2BD38XitFentr8%3D Occurrence Handle11847088
HM Ku T Vision J Liu SD Tanksley (2000) ArticleTitleComparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc Nat Acad Sci USA 97 9121–9126 Occurrence Handle10.1073/pnas.160271297 Occurrence Handle1:CAS:528:DC%2BD3cXls12ks70%3D Occurrence Handle10908680
J Li J Chory (1998) Preparation of DNA from Arabidopsis. JM Martinez-Zapater J Salinas (Eds) Arabidopsis protocols. Humana Press Totowa, NJ 55–60
T MacDonald JL Brewbaker (1974) ArticleTitleIsoenzyme polymorphism in flowering plants. IX. The E5-E10 esterase loci of maize. J Hered 65 37–42 Occurrence Handle1:CAS:528:DyaE2cXksVOmsr0%3D
AG Manganaris FH Alston (1992) ArticleTitleGenetics of esterase isoenzymes in Malus. Theor Appl Genet 83 467–475
Nicholas KB, Nicholas HBJ (1997) GeneDoc: A tool for editing and annotating multiple sequence alignments (version2.6.002). Distributed by the authors (www.psc.edu/biomed/genedoc/ )
JG Oakeshott C Collet RW Phillis KM Nielsen RJ Russell GK Chambers V Ross RC Richmond (1987) ArticleTitleMolecular cloning and characterization of esterase-6, a serine hydrolase of Drosophila. Proc Natl Acad Sci USA 84 3359–3363 Occurrence Handle1:CAS:528:DyaL2sXls12kt7s%3D Occurrence Handle3106966
JG Oakeshott TM Boyce RJ Russell MJ Healy (1995) ArticleTitleMolecular insights into the evolution of an enzyme: Esterase 6 in Drosophila. Trends Ecol Evol 10 103–110 Occurrence Handle10.1016/S0169-5347(00)89003-5
JG Oakeshott C Claudianos RJ Russell GC Robin (1999) ArticleTitleCarboxyl/cholinesterases: A case study of the evolution of a successful multigene family. Bioessays 21 1031–1042 Occurrence Handle10.1002/(SICI)1521-1878(199912)22:1<1031::AID-BIES7>3.3.CO;2-A Occurrence Handle1:STN:280:DC%2BD3c%2FkslOhsg%3D%3D Occurrence Handle10580988
DL Ollis E Cheah M Cygler B Dijkstra F Frolow SM Franken M Harel SJ Remington I Silman J Schrag JL Sussman KHG Verschueren A Goldman (1992) ArticleTitleThe alpha/beta hydrolase fold. Protein Eng 5 197–211 Occurrence Handle1:CAS:528:DyaK38Xlt1OhtL8%3D Occurrence Handle1409539
RDM Page (1996) ArticleTitleTREEVIEW: An application to display phylogenetic trees on personal computers. Comput Appl Biosci 12 357–358 Occurrence Handle1:STN:280:ByiD2MfgtlA%3D Occurrence Handle8902363
SB Petersen F Drablos (1996) A sequence analysis of lipases, esterases and related proteins. P Woolley SB Petersen (Eds) Lipases: Their structure, biochemistry and application. Cambridge University Press Cambridge 23–48
E Pichersky J Gershenzon (2002) ArticleTitleThe formation and function of plant volatiles: Perfumes for pollinator attraction and defense. Curr Opin Plant Biol 5 237–243
D Pontier L Godiard Y Marco D Roby (1994) ArticleTitle hsr203J, a tobacco gene whose activation is rapid, highly localized and specific for incompatible plant/pathogen interactions. Plant J 5 507–521 Occurrence Handle1:CAS:528:DyaK2MXhsl2gsQ%3D%3D Occurrence Handle8012404
D Pontier M Tronchet P Rogowsky E Lam D Roby (1998) ArticleTitleActivation of hsr203, a plant gene expressed during incompatible plant-pathogen interactions, is correlated with programmed cell death. Mol Plant Microbe Interact 11 544–554 Occurrence Handle1:CAS:528:DyaK1cXjsFSmsro%3D Occurrence Handle9612953
H Ranson C Claudianos F Ortelli C Abgrall J Hemingway MV Sharakhova MF Unger FH Collins R Feyereisen (2002) ArticleTitleEvolution of supergene families associated with insecticide resistance. Science 298 179–181 Occurrence Handle10.1126/science.1076781 Occurrence Handle1:CAS:528:DC%2BD38XnsFSgsrg%3D Occurrence Handle12364796
E Richly J Kurth D Leister (2002) ArticleTitleMode of amplification and reorganization of resistance genes during recent Arabidopsis thaliana evolution. Mol Biol Evol 19 76–84 Occurrence Handle1:CAS:528:DC%2BD38XhtFCitg%3D%3D Occurrence Handle11752192
PJ Roy JM Stuart J Lund SK Kim (2002) ArticleTitleChromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418 975–979 Occurrence Handle10.1038/nature01012 Occurrence Handle1:CAS:528:DC%2BD38XmsFWkt78%3D Occurrence Handle12214599
RJ Russell GC Robin P Kostakos RD Newcomb TM Boyce KM Medveczky JG Oakeshott (1996) ArticleTitleMolecular cloning of an alpha-esterase gene cluster on chromosome 3r of Drosophila melanogaster. Insect Biochem Mol Biol 26 235–247 Occurrence Handle10.1016/0965-1748(95)00081-X Occurrence Handle1:CAS:528:DyaK28XitVKrtrc%3D Occurrence Handle8900595
H Sandermann Jr (1992) ArticleTitlePlant metabolism of xenobiotics. Trends Biochem Sci 17 82–84 Occurrence Handle10.1016/0968-0004(92)90507-6 Occurrence Handle1:STN:280:By2B38bot1Y%3D Occurrence Handle1566333
T Satoh M Hosokawa (1998) ArticleTitleThe mammalian carboxylesterases: From molecules to functions. Annu Rev Pharmacol Toxicol 38 257–288
H Schoof P Zaccaria H Gundlach K Lemcke S Rudd G Kolesov R Arnold HW Mewes KF Mayer (2002) ArticleTitleMIPS Arabidopsis thaliana Database (MAtDB): An integrated biological knowledge resource based on the first complete plant genome. Nucleic Acids Res 30 91–93 Occurrence Handle10.1093/nar/30.1.91 Occurrence Handle1:CAS:528:DC%2BD38Xht12kt70%3D Occurrence Handle11752263
C Simillion K Vandepoele MC Van Montagu M Zabeau Y Van De Peer (2002) ArticleTitleThe hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci USA 99 13627–13632 Occurrence Handle10.1073/pnas.212522399 Occurrence Handle1:CAS:528:DC%2BD38XotVKmsbY%3D Occurrence Handle12374856
PT Spellman GM Rubin (2002) ArticleTitleEvidence for large domains of similarly expressed genes in the Drosophila genome. J Biol 1 5.1–5.8 Occurrence Handle10.1186/1475-4924-1-5
R Stracke M Werber B Weisshaar (2001) ArticleTitleThe R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol 4 447–456 Occurrence Handle10.1016/S1369-5266(00)00199-0 Occurrence Handle1:CAS:528:DC%2BD3MXmvFKgt74%3D Occurrence Handle11597504
C Stuhlfelder F Lottspeich MJ Mueller (2002) ArticleTitlePurification and partial amino acid sequences of an esterase from tomato. Phytochemistry 60 233–240 Occurrence Handle10.1016/S0031-9422(02)00126-7 Occurrence Handle1:CAS:528:DC%2BD38XjvVakuro%3D Occurrence Handle12031441
JL Sussman M Harel F Frolow C Oefner A Goldman L Toker I Silman (1991) ArticleTitleAtomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 253 872–879 Occurrence Handle1:CAS:528:DyaK3MXlslyitrc%3D Occurrence Handle1678899
SD Tanksley CM Rick (1980) ArticleTitleGenetics of esterases in species of Lycopersicon. Theor Appl Genet 56 209–219 Occurrence Handle1:CAS:528:DyaL3cXhslOmt7c%3D
P Taylor Z Radic (1994) ArticleTitleThe cholinesterases: From genes to proteins. Annu Rev Pharmacol Toxicol 34 281–320 Occurrence Handle1:CAS:528:DyaK2cXltVamsLo%3D Occurrence Handle8042853
InstitutionalAuthorNameThe Arabidopsis Genome Initiative (2000) ArticleTitleAnalysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 796–815 Occurrence Handle11130711
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res 24 4876–4882 Occurrence Handle10.1093/nar/25.24.4876
M Tronchet B Ranty Y Marco D Roby (2001) ArticleTitle HSR203 antisense suppression in tobacco accelerates development of hypersensitive cell death. Plant J 27 115–127 Occurrence Handle10.1046/j.1365-313x.2001.01072.x Occurrence Handle1:CAS:528:DC%2BD3MXmsVyjtrs%3D Occurrence Handle11489189
JM Turner NA Larsen A Basran CF, 3r Barbas NC Bruce IA Wilson RA Lerner (2002) ArticleTitleBiochemical characterization and structural analysis of a highly proficient cocaine esterase. Biochemistry 41 12297–12307 Occurrence Handle10.1021/bi026131p Occurrence Handle1:CAS:528:DC%2BD38XntFejtL0%3D Occurrence Handle12369817
TJ Vision DG Brown DB Shmoys RT Durrett SD Tanksley (2000) ArticleTitleSelective mapping: A strategy for optimizing the construction of high-density linkage maps. Genetics 155 407–420 Occurrence Handle1:STN:280:DC%2BD3c3ltlWhsA%3D%3D Occurrence Handle10790413
RG Vogt LM Riddiford GD Prestwich (1985) ArticleTitleKinetic properties of a sex pheromone-degrading enzyme: The sensillar esterase of Antheraea polyphemus. Proc Natl Acad Sci USA 82 8827–8831 Occurrence Handle1:CAS:528:DyaL28XhtVCqurc%3D Occurrence Handle3001718
AR Walden C Walter RC Gardner (1999) ArticleTitleGenes expressed in Pinus radiata male cones include homologs to anther-specific and pathogenesis response genes. Plant Physiol 121 1103–1116 Occurrence Handle10.1104/pp.121.4.1103 Occurrence Handle1:CAS:528:DyaK1MXotFyju74%3D Occurrence Handle10594098
Acknowledgements
We gratefully acknowledge Julie Nicolls for her assistance in glasshouse work and Revel Drummond for help with cDNA preparation. S.D.G.M. is the recipient of a Bright Futures Enterprise Scholarship from the Foundation for Research, Science and Technology (New Zealand). Funding for this work has been provided by The Foundation for Research, Science and Technology (C06X0207).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marshall, S.D., Putterill, J.J., Plummer, K.M. et al. The Carboxylesterase Gene Family from Arabidopsis thaliana . J Mol Evol 57, 487–500 (2003). https://doi.org/10.1007/s00239-003-2492-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2492-8