
Abstract
Continuous in vitro evolution methods were used to study the behavior of an evolving population of RNA ligase ribozymes in response to selection pressures involving conditions of extreme pH. The starting population consisted of randomized variants of a ribozyme that had been optimized for activity at pH 8.5. The ribozymes were subjected to repeated rounds of selective amplification under progressively more acidic or more alkaline conditions. The two final evolved populations of ribozymes were able to operate at either pH 5.8 or pH 9.8, respectively. Representative individuals from the two final populations were isolated and characterized. The low-pH ribozyme exhibited a 10-fold increase in catalytic rate at pH 5.8 compared to the starting molecule. The high-pH ribozyme retained its structural integrity and activity at pH 9.8, whereas the starting molecule was denatured under this condition. These findings demonstrate that a population of functional macromolecules can adapt to stringent environmental conditions through the acquisition of relatively few mutations. The results establish continuous in vitro evolution as a useful model system for exploring the evolution of enzymatic function in extreme environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The “RNA world” hypothesis (Gilbert 1986; Joyce 2002) postulates that RNA-based life preceded DNA- and protein-based life, with RNA acting as both the carrier of genetic information and the principal agent of catalytic function. As environmental conditions changed in the RNA world, catalytic RNA molecules (ribozymes) would have adapted accordingly, resulting in the evolution of RNAs with altered functional properties. In vitro RNA evolution provides a model of RNA evolution in the RNA world. The experimenter can construct a heterogeneous population of catalytic RNA molecules and subject them to defined selection pressures, resulting in the evolution of molecules that have adapted to those pressures. Most in vitro evolution experiments focus on the development of a particular catalytic function, rather than the response of an evolving population to changing environmental conditions. Two notable exceptions are the work of Schmitt and Lehman (1999), in which a ligase ribozyme was evolved to operate in the presence of progressively lower concentrations of MgCl2, and the work of Ito and co-workers (Miyamoto et al. 2001), in which a similar ligase ribozyme was evolved to operate at acidic pH.
Nature provides many examples of organisms that have adapted to extreme environmental conditions. The acidophilic bacterium Picrophilus oshimae lives in acidic hot springs at pH < 3 (Schleper et al. 1995), while the alkalophilic bacterium Natronobacterium magadi thrives in soda lakes at pH > 10 (Kroll 1990). Thermophilic bacteria, such as Thermus aquaticus, can survive in volcanic hot springs, while the psychrophilic bacterium Polaromonas vacuolata can withstand the extreme cold of Antarctic fellfields (Madigan and Marrs 1997). The complement of enzymes within these organisms also must have adapted to function at extremes of pH and temperature. This adaptation might involve changes in the biophysical properties of the enzymes themselves or alterations of the cellular environment that mitigate the effects of the external environment. The polymerase enzymes are especially important to the survival of biological organisms because they are required for copying the genetic information. Polymerases have been isolated from extremophiles and have been the subject of intense investigation because of their biochemical interest and potential applications in biotechnology.
The present study sought to explore the evolution of ribozymes with polymerase-like activity under controlled laboratory conditions involving extremes of either low or high pH. The starting point for these studies was an in vitro evolved form of the class I ligase ribozyme, first isolated almost 10 years ago (Bartel and Szostak 1993; Ekland et al., 1995). This ribozyme catalyzes the template-directed joining of an oligonucleotide to its own 5′ end through formation of a 3′, 5′-phosphodiester linkage. It also catalyzes the polymerization of up to three nucleoside 5′-triphosphates (NTPs) on an internal RNA template (Ekland and Bartel 1996) and was further evolved to catalyze the polymerization of up to 14 NTPs on an external RNA template (Johnston et al. 2001). Thus the class I ligase and its evolutionary descendants can be viewed as representative of an RNA-dependent RNA polymerase, an activity that would have been required for the replication of RNA genomes in the RNA world.
The class I ligase ribozyme originally was selected to operate in the presence of 60 mM MgCl2 and 600 mM KCl at pH 7.4 and 25°C (Bartel and Szostak 1993; Ekland et al. 1995). The in vitro evolution scheme that led to its development involved challenging a population of RNA molecules to react with an oligonucleotide substrate that contained a tag sequence at its 5′ end. All of the RNA molecules in the population then were reverse transcribed, but only those that had performed the reaction gave rise to a complementary DNA that contained the complement of the tag sequence. This sequence served as a primer-binding site for PCR amplification, resulting in selective amplification of only those DNA sequences that corresponded to reacted ribozymes. The PCR products were transcribed to produce a “progeny” population of RNA molecules, which could be used to initiate another round of selective amplification.
This “stepwise” method for in vitro evolution contrasts with “continuous” in vitro evolution methods that were developed more recently (Wright and Joyce 1997) and provide a more realistic model of Darwinian evolution in nature. In continuous evolution, RNA catalysis and selective amplification take place within a common reaction mixture, such that the reacted molecules immediately are eligible for amplification and the newly synthesized progeny molecules can immediately perform another reaction. Continuous evolution can be carried out indefinitely by employing a serial-transfer procedure, involving periodic transfer of a small portion of a completed reaction mixture to a new reaction vessel that contains a fresh supply of the substrate and other materials necessary for catalysis and selective amplification.
Continuous in vitro evolution methods have been applied to the class I ligase ribozyme (Fig. 1). The ribozyme first was evolved in a stepwise manner to operate under conditions necessary for amplification by a combination of reverse transcriptase and T7 RNA polymerase. It also was evolved to react with an oligonucleotide substrate that contained the sequence of the T7 RNA polymerase promoter, so that reverse transcripts of reacted, but not unreacted, ribozymes would serve as templates for forward transcription (Wright and Joyce 1997). The first continuous in vitro evolution experiment was initiated with a population of ˜10 pmol of ligase ribozyme, which was carried through 100 successive transfers corresponding to an overall dilution of 3 × 10298-fold. This resulted in a variant form of the class I ligase, termed “E100-3,” that operated with a catalytic efficiency (k cat/K m) of 1 × 107 M −1 min−1 in the presence of 15 mM MgCl2 and 50 mM KCl at pH 8.5 and 37°C (Wright and Joyce 1997).

Scheme for continuous in vitro evolution of RNA ligase ribozymes. The ribozyme reacts with a chimeric DNA–RNA substrate that contains the sequence of the T7 RNA polymerase promoter [prom −], joining the substrate to its 5′ end and releasing inorganic pyrophosphate. A DNA primer binds to the 3′ end of the ribozyme and is extended by reverse transcriptase, continuing through the promoter sequence only if the ribozyme has performed the reaction. The functional double-stranded promoter is recognized by T7 RNA polymerase, which generates multiple copies of RNA per copy of DNA template. The “progeny” RNAs can enter another cycle of reaction and selective amplification. DNA and RNA are shown as gray and black lines, respectively (figure adapted from Wright and Joyce [1997]).
The El00-3 ribozyme has been used as a starting point for other continuous in vitro evolution experiments. In one case, it was evolved to be resistant to attack by an RNA-cleaving DNA enzyme. The ribozyme accomplished this by blocking binding of the DNA enzyme and by increasing affinity for its own substrate (Ordoukhanian and Joyce 1999). In another study, the ribozyme was evolved to operate in the presence of progressively lower concentrations of Mg2+, ultimately requiring the molecule to form a properly folded structure in the presence of only 3.7 mM free Mg2+ (Schmitt and Lehman 1999). In yet another study, the ribozyme was evolved to perform three successive nucleotidyl addition reactions: two template-directed NTP additions followed by RNA ligation (McGinness et al. 2002).
The present study sought to explore the limits of ribozyme function under conditions of extreme pH, again employing continuous in vitro evolution starting with the El00-3 ligase. At low pH the rate of the chemical step of the reaction is slowed considerably (Bergman et al. 2000), imposing selection pressure favoring an increase in the catalytic rate. At high pH the secondary and tertiary structure of RNA is destabilized, imposing selection pressure favoring enhancement of the molecule’s structural integrity. The appropriate evolutionary responses were observed in both cases, illustrating how environmental factors govern ribozyme function and how ribozymes can adapt to novel environmental conditions by acquiring a modest number of critical mutations.
Materials and Methods
Materials
NaCl, KCl, MgCl2, Na2HPO4, KH2PO4, spermidine, dithiothreitol (DTT), ethylenediaminetetraacetic acid (EDTA), 2-[N-morpholino]ethanesulfonic acid (MES), N-[2-hydroxyethyl]piperazine-N′-[3-propanesulfonic acid] (EPPS), and 2-[N-cyclohexylamino] ethanesulfonic acid (CHES) were purchased from Sigma–Aldrich. Synthetic oligonucleotides were prepared by automated synthesis using an Applied Biosystems Expedite nucleic acid synthesizer, then purified by denaturing polyacrylamide gel electrophoresis. Histidine-tagged T7 RNA polymerase was purified from E. coli strain BL21 containing plasmid pBH161 (kindly provided by William McAllister), using His-Bind resin (Novagen) according to the manufacturer’s protocol. Thermus aquaticus DNA polymerase was cloned from total genomic DNA and purified as previously described (Pluthero 1993). Superscript and Stratascript RNase H− reverse transcriptase were purchased from Life Technologies and Stratagene, respectively. Nucleoside 5′-triphosphates (NTPs) and deoxynucleoside 5′-triphosphates (dNTPs) were from USB/Amersham-Pharmacia and [α32-P]ATP was from ICN Radiochemicals. Cycle sequencing reagents, including ThermoSequenase and dideoxynucleotides, were from USB/Amersham–Pharmacia.
In Vitro Evolution
Cloned DNA encoding the E100-3 RNA ligase (Wright and Joyce 1997) was PCR amplified, purified by agarose gel electrophoresis, and subjected to mutagenic PCR that introduced mutations at a frequency of 10% per nucleotide position (Vartanian et al. 1996). The resulting DNAs were transcribed in vitro to generate the starting pool of RNAs. The RNAs were purified by denaturing polyacrylamide gel electrophoresis, and 1 pmol of the purified material was used to initiate continuous in vitro evolution. Continuous evolution was carried out as previously described (Wright and Joyce 1997), except that different buffers were employed at low and high pH: MES was used at pH 5.8–7.2, EPPS at pH 7.2–8.8, and CHES at pH 8.8–9.8. In addition to the ligase ribozyme, the continuous evolution mixture contained 5 µM substrate having the sequence 5′-CTTGACGTCCAGCCTGGACTAATACGACTCACUAUA-3′ (promoter sequence underlined; RNA residues in boldface), 2 µM reverse transcription primer having the sequence 5′-GCTGAGCCTGCGATTGG-3′, 25 mM MgCl2, 50 mM KCl, 2 mM spermidine, 5 mM DTT, 50 mM buffer, 200 µM of each of the four dNTPs, 2 mM of each of the four NTPs, 200 U µl−1 Superscript or 50 U µl−1 Stratascript reverse transcriptase, and 100 U µl−1 T7 RNA polymerase, which were incubated at 37°C for 1–2 h. A small aliquot of the mixture then was transferred to a fresh reaction vessel containing all of the above components. Dilutions between successive transfers were either 100-fold, for the early rounds, or 10-fold for the later rounds.
Kinetic Analysis
Ligation reactions were carried out under substrate-excess conditions, employing 1–20 nM [α32-P]ATP-labeled ribozyme, 0.2–5 µM unlabeled substrate, 25 mM MgCl2, 50 mM KC1, and 60 mM buffer at 37°C. The reaction products were separated by denaturing polyacrylamide gel electrophoresis and the amount of reacted and unreacted ribozyme was determined using a PhosphorImager (Molecular Dynamics). Data were analyzed using Kaleidagraph. The maximum extent of the reaction was determined from long time points. Semilogarithmic plots of the fraction unreacted versus time were used to determine the apparent rate constant for each concentration of substrate. Values for k cat and K m were obtained from an Eadie–Hofstee plot.
Thermal Denaturation
Samples for thermal-denaturation analysis were diluted in 150 mM NaCl, 5 mM Na2HPO4/5 mM KH2PO4 (pH 8.0), and 0.1 mM Na2EDTA to achieve an initial absorbance of 0.35 AU at 254 nm (˜250 nM ribozyme). The samples were degassed in a vacuum centrifuge for 10 min, topped off to their original volume with degassed double-distilled H2O, then equilibrated at 10°C. Thermal-denaturation measurements were carried out using a Varian Cary 1 Bio UV-visible spectrophotometer fitted with a temperature-controlled multiple cell holder. Data were obtained at 0.5°C intervals, ramping forward and backward several times from 10 to 95°C at a ramping speed of 1.0°C min−1. The data were analyzed using PRISM (with permission from G. W. Brudvig) and Kaleidagraph.
Results
Continuous In Vitro Evolution
The E100-3 RNA ligase originally was developed to undergo continuous evolution in the presence of 25 mM MgCl2 at pH 8.5 and 37°C (Wright and Joyce 1997). This molecule was subjected to random mutagenesis (Vartanian et al. 1996), resulting in a heterogeneous population of ribozyme variants. The randomized population could be propagated at pH 8.5 by a serial transfer protocol involving successive 2-h incubations and 10-fold dilutions. It similarly could be propagated at any pH in the range of 6.4–9.2 but could not survive at pH < 6.4 or pH > 9.2.
Two evolutionary lineages were initiated, one starting at pH 6.4 and the other at pH 9.2, then gradually lowering or raising the pH to explore more extreme conditions. The low-pH lineage was subjected to 66 serial transfers, involving a total reaction time of 79 h and an overall dilution of 1075-fold. During this time the pH of the reaction mixture was reduced progressively from 6.4 to 5.8. Random mutagenesis was carried out following the 18th and 48th transfers, introducing mutations at a frequency of 10% and 0.7% per nucleotide position, respectively (Cadwell and Joyce 1992; Vartanian et al. 1996). The high-pH lineage was subjected to 47 serial transfers, involving a total reaction time of 61 h and an overall dilution of 1050-fold, during which the pH was raised from 9.2 to 9.8. Random mutagenesis was carried out following the 18th and 28th transfers, introducing mutations at a frequency of 10% per nucleotide position in both instances. Below pH 5.8 and above pH 9.8 the activity of the two polymerase proteins used to carry out RNA amplification was less than 5% of their activity level at pH 8.5, preventing continuous evolution from operating.
Sequence Analysis of Evolved Ribozymes
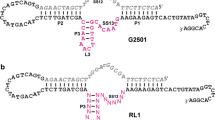
Individual clones were isolated from the two final evolved populations, sequenced, and tested for catalytic activity. Ten clones were examined for each lineage, revealing some mutations that were present in all cases and others that were confined to either the low-pH or the high-pH lineage (Fig. 2). Most of the commonly occurring mutations have been seen in other continuous in vitro evolution experiments that were initiated with the E100-3 ligase ribozyme (Fig. 2, gray circles) (Schmitt and Lehman 1999; Ordoukhanian and Joyce 1999; McGinness et al. 2002). Ten of these mutations involved replacement of a base pair within a stem region by a different pair of complementary nucleotides, and five involved a single-nucleotide substitution or deletion within an unpaired region. These common mutations appear to confer a general selective advantage within the context of continuous in vitro evolution.

Composition of the starting ligase ribozyme and variants that were evolved to operate under conditions of either low or high pH. a The starting E100-3 ribozyme (Wright and Joyce 1997). b The 5.8-12 ribozyme, which operates at low pH. c The 9.8-15 ribozyme, which operates at high pH. Mutations that were common to these and other continuous in vitro evolution experiments are indicated by gray circles; those that were specific to either the low-pH or the high-pH lineage are indicated by black circles; the remainder are indicated by open circles. Deletions relative to the E100-3 ribozyme are indicated by a dash. Mutated positions are numbered. Open rectangles correspond to the 5′ portion of the substrate and the fixed primer binding site at the 3′ end of the ribozyme (see Materials and Methods).
Surprisingly, residue U12, which is thought to form a Watson–Crick pair with the 5′-terminal nucleotide of the ribozyme, was deleted in some of the individuals isolated from both the low-pH and the high-pH lineages (Fig. 2). This mutation has not been seen in any previous in vitro evolution experiment. Other mutations were observed exclusively in either the low-pH or the high-pH lineage (Fig. 2, black circles). All of the high-pH clones, for example, contained an A → C change at position 11, which was not present in the low-pH lineage. All of the high-pH clones also contained an insertion of several nucleotides located near the 3′ end of the molecule. Mutations that became fixed within only one of the two lineages are expected to confer enhanced activity under the corresponding set of environmental conditions. However, some of these mutations may be neutral with regard to phenotype, merely accompanying other mutations that provide a selective advantage.
Kinetic Analysis
A representative cloned individual was chosen from each of the two lineages for more detailed kinetic characterization. The chosen individuals from the low-pH and high-pH lineages were designated “5.8-12” and “9.8-15,” respectively (Fig. 2b and 2c). Their behavior was compared to that of the starting E100-3 ligase ribozyme. The observed rate of ligation was determined for a range of substrate concentrations, and these data were used to calculate k cat and K m (Fig. 3, Table 1).

Lineweaver–Burk plots of representative kinetic data for the two evolved ribozymes. a The 5.8-12 ribozyme operating at pH 5.8, employing 1 nM ribozyme and 0.2–5 µM substrate. b The 9.8-15 ribozyme operating at pH 9.5, employing 10 nM ribozyme and 0.2–5 µM substrate. Individual values for v 0 were obtained over the first 2 min of the reaction.
All three ribozymes exhibited comparable kinetic behavior at pH 8.4, with a k cat of 10–20 min−1 and a K m of 2–5 µM. At pH 5.8, however, the catalytic rate of the E100-3 ribozyme was reduced by about 50-fold, while that of the 5.8-12 ribozyme was reduced by only 3-fold. At pH 9.5 the E100-3 ribozyme became denatured (see below) and did not exhibit any appreciable catalytic activity. The [α-32P]-labeled ribozyme appeared as a mixture of degradation products when examined by polyacrylamide gel electrophoresis (data not shown). Despite the significant extent of RNA degradation, it was possible to detect catalytic activity of the E100-3 ribozyme up to pH 9.1. However, its activity was significantly impaired under these conditions, presumably due to partial denaturation and/or degradation. In contrast, the 9.8-15 ribozyme retained sufficient structural integrity at elevated pH that its kinetic properties could be determined at pH 9.5, revealing a k cat of 2.4 ± 0.8 min−1 and a K m of 3.3 ± 1.1 µM. Activity of the 9.8-15 ribozyme was detected at pH 9.8 but could not be measured accurately due to competing degradation of the RNA.
Thermal Stability
Thermal denaturation experiments were carried out to compare the structural stability of the E100-3 and 9.8-15 ribozymes. Ultraviolet absorbance was monitored while repeatedly cycling the temperature between 10 and 95°C. This provided well-behaved and highly reproducible thermal-denaturation curves, from which the first derivative was determined to emphasize transitions between the folded and the unfolded states (Fig. 4). The E100-3 ribozyme exhibited a major peak at ˜38°C, corresponding to an increase in absorbance due to destabilization of its helical structure. The 9.8-15 ribozyme exhibited two peaks, one at ˜38°C and another at ˜58°C (Fig. 4). This finding suggests either that the RNA becomes partially unfolded at the lower transition temperature and then fully unfolded at the higher transition temperature or that it can adopt two different conformations, one that unfolds at the same temperature as the E100-3 ribozyme and another that remains in the folded state up to approximately 58°C. These experiments were performed in the absence of MgCl2 to prevent degradation of the RNA, which may lower the transition temperature compared to when MgCl2 is present.

First derivative of the thermal denaturation profile for the E100-3 and 9.8-15 ribozymes. Ultraviolet absorbance was monitored at 254 nm while cycling the temperature between 10 and 95°C, at a ramping speed of 1.0°C min−1 (see Materials and Methods). The data were smoothed by averaging each point with the three adjacent points on either side and scaled to unity. The traces in the figure represent data from an individual melting event.
The denaturing effects of elevated pH and elevated temperature are not strictly comparable, although they tend to be highly correlated. The thermal denaturation experiments were conducted at pH 8.0 and could not be carried out above pH 9.0 due to substantial degradation of the RNA during the repeated heating cycles. In order to examine the ability of the E100-3 and 9.8-15 ribozymes to operate under partially denaturing conditions, comparative kinetic analyses were performed at pH 8.0 and 42°C. Under these conditions the E100-3 ribozyme exhibited a k cat of 0.94 ± 0.04 min−1, which is more than 10-fold lower than its catalytic rate at pH 8.4 and 37°C. In contrast, the 9.8-15 ribozyme exhibited a k cat of 4.3 ± 2.2 min−1 at pH 8.0 and 42°C. Thus the ability of the evolved 9.8-15 ribozyme to operate more efficiently at elevated pH also confers greater thermostability compared to the starting E100-3 ribozyme.
Discussion
Only a few mutations were necessary to bring about enhanced activity of the evolved ribozymes under either low-pH or high-pH conditions. Some of these mutations have been observed in other continuous in vitro evolution experiments involving the E100-3 ligase, suggesting that they confer general improvement in the fitness of the ribozyme (Fig. 2, gray circles). Other mutations appear to be specific to either of the two conditions of extreme pH, resulting in either improved catalytic rate at low pH or enhanced structural integrity at high pH (Fig. 2, black circles). It is unlikely that the small number of pH-specific mutations cause a major remodeling of the global structure of the ribozyme. More likely, they subtly perturb a few critical residues, helping to offset a difference of four pH units between the two extreme conditions. Structural stabilization at high pH did not involve a significant increase in the length or GC content of the stem regions of the ribozyme. The inserted nucleotides at the 3′ end of the 9.8-15 ligase may provide additional tertiary contacts that help stabilize the overall structure, but the nature of these contacts is not obvious. Similarly, comparisons of naturally occurring protein enzymes that have been adapted to either moderate or extreme temperatures reveal only subtle differences in their overall structure (Eom et al. 1996; Knapp et al. 1997; Russell et al. 1998).
Bartel and co-workers have studied the pH dependence of the prototype class I ligase ribozyme (Bergman et al. 2000). Its catalytic rate increases log-linearly with increasing pH over the range of 5.7–7.0. Above pH 7.0 the observed rate increases more slowly, presumably because it is limited by something other than the chemical step of the reaction. The K m of the class I ligase is significantly greater at alkaline compared to neutral pH, consistent with reduced substrate-binding affinity under alkaline conditions. The kinetic properties of this ribozyme and the derived E100-3 ribozyme are similar but not strictly comparable due to differences in their corresponding substrates and preferred reaction conditions. Both ribozymes react to a maximum extent of substantially less than 100%. For the class I ligase, this has been attributed to a minor fraction (˜15%) that has lost its 5′-triphosphate and thus is unable to react, and another fraction (˜15%) that is trapped in a slow-reacting conformation (Bergman et al. 2000). Similar limitations exist for the ribozymes of the present study, especially under conditions of extreme pH.
In a recent study by Ito and co-workers (Miyamoto et al. 2001), a slight variant of the class I ligase (containing one mutation found in the E100-3 ligase) was subjected to four rounds of “stepwise” in vitro evolution, selecting for catalytic activity at pH 4.0. A representative individual from the final selected population contained four mutations relative to the starting ribozyme and exhibited a 400-fold improvement in catalytic rate at pH 4.0. Its observed rate was about 3 × 10−5 min−1 at pH 4.0 and about 3 × 10−2 min−1 at pH 6.0. None of the four mutations that it contained were present in the 5.8-12 ribozyme of the present study. One of these mutations, however, was an 11-nucleotide insertion near the 3′ end that was similar in location but not in sequence to eight nucleotides that were inserted near the 3′ end of the 9.8-15 ribozyme (Fig. 2c). The low-pH ribozyme obtained by Miyamoto et al. (2001) is about 100-fold slower at pH 6.0 compared to either the class I ligase (Bergman et al. 2000) or the 5.8-12 ligase but appears to have relatively better performance at pH 4.0.
The present study employed continuous in vitro evolution methods to subject a population of RNA ligase ribozymes to dozens of rounds of selective amplification under conditions of progressively more extreme pH. While continuous evolution is a powerful technique, it is limited by the range of conditions that are tolerated by the protein polymerases needed for RNA amplification. This range could be expanded if reverse transcriptase and RNA polymerase enzymes were available that could operate efficiently below pH 5.8 or above pH 9.8. Similarly, the adaptation of biological organisms to progressively more extreme environmental conditions requires that the replication machinery adapt to those conditions. Even if it were possible to design a metabolic enzyme that could function in an extreme environment, such an enzyme might not be obtainable through cell-based Darwinian evolution.
References
DP Bartel JW Szostak (1993) ArticleTitleIsolation of new ribozymes from a large pool of random sequences. Science 261 1411–1418 Occurrence Handle7690155
NH Bergman WK Johnston DP Bartel (2000) ArticleTitleKinetic framework for ligation by an efficient RNA ligase ribozyme. Biochemistry 39 3115–3123 Occurrence Handle10.1021/bi992654u Occurrence Handle10715133
RC Cadwell GF Joyce (1992) ArticleTitleRandomization of genes by PCR mutagenesis. PCR Methods Appl 2 28–33 Occurrence Handle1:CAS:528:DyaK3sXhs1Sr Occurrence Handle1490172
EH Ekland JW Szostak DP Bartel (1995) ArticleTitleStructurally complex and highly active RNA ligases derived from random RNA sequences. Science 269 364–370 Occurrence Handle7618102
EH Ekland DP Bartel (1996) ArticleTitleRNA-catalysed RNA polymerization using nucleoside triphosphates. Nature 382 373–376 Occurrence Handle1:CAS:528:DyaK28Xks1Wnsrg%3D Occurrence Handle8684470
SH Eom J Wang TA Steitz (1996) ArticleTitleStructure of Taq polymerase with DNA at the polymerase active site. Nature 382 278–281 Occurrence Handle10.1038/382278a0 Occurrence Handle8717047
W Gilbert (1986) ArticleTitleThe RNA world. Nature 319 618
WK Johnston PJ Unrau MS Lawrence ME Glasner DP Bartel (2001) ArticleTitleRNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science 292 1319–1325 Occurrence Handle1:CAS:528:DC%2BD3MXjvVGjsbg%3D Occurrence Handle11358999
GF Joyce (2002) ArticleTitleThe antiquity of RNA-based evolution. Nature 418 214–221 Occurrence Handle10.1038/418214a Occurrence Handle12110897
S Knapp WM de Vos D Rice R Ladenstein (1997) ArticleTitleCrystal structure of glutamate dehydrogenase from the hyperthermophilic eubacterium Thermatoga maritima at 3.0 Å resolution. J Mol Biol 267 916–932 Occurrence Handle1:CAS:528:DyaK2sXivFWks7w%3D Occurrence Handle9135121
RG Kroll (1990) Alkalophiles. EC Edwards (Eds) Microbiology of extreme environments. Open University Press Milton Keynes, UK 55–92
MT Madigan BL Marrs (1997) ArticleTitleExtremophiles. Sci Am 1997 82–87
KE McGinness MC Wright GF Joyce (2002) ArticleTitleContinuous in vitro evolution of a ribozyme that catalyzes three successive nucleotidyl addition reactions. Chem Biol 9 585–596 Occurrence Handle10.1016/S1074-5521(02)00136-9 Occurrence Handle12031665
Y Miyamoto N Teramoto Y Imanishi Y Ito (2001) ArticleTitleIn vitro adaptation of a ligase ribozyme for activity under low-pH condition. Biotechnol Bioeng 75 590–596 Occurrence Handle10.1002/bit.10033 Occurrence Handle11745135
P Ordoukhanian GF Joyce (1999) ArticleTitleA molecular description of the evolution of resistance. Chem Biol 6 881–889 Occurrence Handle1:CAS:528:DyaK1MXotVygsro%3D Occurrence Handle10631516
FG Pluthero (1993) ArticleTitleRapid purification of high-activity Taq DNA polymerase. Nucleic Acids Res 21 4850–4851 Occurrence Handle8233838
RJM Russell U Gerike MJ Danson DW Hough GL Taylor (1998) ArticleTitleStructural adaptations of the cold-active citrate synthase from an Antarctic bacterium. Structure 6 351–361 Occurrence Handle1:CAS:528:DyaK1cXisFSls7w%3D
C Schleper G Pühler B Kühlmorgen W Zillig (1995) ArticleTitleLife at extremely low pH. Nature 375 741–742 Occurrence Handle10.1038/375741b0
T Schmitt N Lehman (1999) ArticleTitleNon-unity molecular heritability demonstrated by continuous evolution in vitro. Chem Biol 6 857–869 Occurrence Handle10.1016/S1074-5521(00)80005-8 Occurrence Handle10631514
J-P Vartanian M Henry S Wain-Hobson (1996) ArticleTitleHypermutagenic PCR involving all four transitions and a sizeable proportion of transversions. Nucleic Acids Res 24 2627–2631 Occurrence Handle10.1093/nar/24.14.2627 Occurrence Handle8758987
MC Wright GF Joyce (1997) ArticleTitleContinuous in vitro evolution of catalytic function. Science 276 614–617 Occurrence Handle1:CAS:528:DyaK2sXivVKis7c%3D Occurrence Handle9110984
Acknowledgements
This work was supported by Grant NAG5-9386 from the National Aeronautics and Space Administration and The Skaggs Institute for Chemical Biology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kühne, H., Joyce, G.F. Continuous In Vitro Evolution of Ribozymes That Operate Under Conditions of Extreme pH . J Mol Evol 57, 292–298 (2003). https://doi.org/10.1007/s00239-003-2480-z
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2480-z