
Abstract
Recent analyses have shown that nonsynonymous variation in human mitochondrial DNA (mtDNA) contains nonneutral variants, suggesting the presence of mildly deleterious mutations. Many of the disease-causing mutations in mtDNA occur in the genes encoding the tRNAs. Nucleotide sequence variation in these genes has not been studied in human populations, nor have the structural consequences of nucleotide substitutions in tRNA molecules been examined. We therefore determined the nucleotide sequences of the 22 tRNA genes in the mtDNA of 477 Finns and, also, obtained 435 European sequences from the MitoKor database. No differences in population polymorphism indices were found between the two data sets. We assessed selective constraints against various tRNA domains by comparing allele frequencies between these domains and the synonymous and nonsynonymous sites, respectively. All tRNA domains except the variable loop were more conserved than synonymous sites, and T stem and D stem were more conserved than the respective loops. We also analyzed the energetic consequences of the 96 polymorphisms recovered in the two data sets or in the Mitomap database. The minimum free energy (ΔG) was calculated using the free energy rules as implemented in mfold version 3.1. The ΔG’s were normally distributed among the 22 wild-type tRNA genes, whereas the 96 polymorphic tRNAs departed significantly from a normal distribution. The largest differences in ΔG between the wild-type and the polymorphic tRNAs in the Finnish population tended to be in the polymorphisms that were present at low frequencies. Allele frequency distributions and minimum free energy calculations both suggested that some polymorphisms in tRNA genes are nonneutral.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human mitochondrial genome (mtDNA) is a double-stranded, circular molecule of 16 569 nucleotide pairs, and each mitochondrion contains several identical copies of mtDNA. This genome is maternally transmitted and does not undergo recombination that is detectable at a population level (Piganeau and Eyre-Walker 2004). MtDNA encodes 13 of the more than 80 polypeptides in the mitochondrial respiratory-chain complexes and contains genes for 2 rRNAs and 22 tRNAs required for mitochondrial protein synthesis. There are several copies of each tRNA gene in the nuclear DNA, but there is only one copy of each tRNA gene in mtDNA. The rate of new mutations in mtDNA is approximately 10 to 17 times greater than that in the nuclear genome (Neckelmann et al. 1987; Wallace et al. 1997), but the substitution rate in mtDNA varies between regions, so that the tRNA genes, for example, evolve much more slowly than synonymous sites (Pesole et al. 1999).
A comparative study of the tRNA genes in animal mitochondrial and nuclear genomes has demonstrated that the former accumulate nucleotide substitutions much more rapidly than do the latter, and several lines of evidence are consistent with the idea that the excess substitutions are mildly deleterious mutations (Lynch 1996). Such mutations would give rise to a class of haplotypes that are kept at low frequency by their mildly deleterious effects but not eliminated from the population immediately (Fry 1999). Mildly deleterious mutations would be compatible with life, but mitochondrial function would decline with age in postmitotic tissues and lead to OXPHOS deficiency and ultimately to clinical symptoms (Wallace 1995). Many of the disease-causing mutations in mtDNA occur in the genes encoding tRNA, where the greatest number of pathogenic mutations have been identified in the MTTL1 gene, encoding tRNALeu(UUR). The heteroplasmic mutation 3243A>G in this gene, for example, leads to a decrease in the synthesis of mtDNA-encoded proteins and causes the MELAS syndrome (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes).
Many disease-causing point mutations have already been identified in mtDNA, but there is evidence to suggest that the number of mutations and polymorphisms is probably higher than is currently known (Moilanen and Majamaa 2003). Nucleotide sequence variation in the mtDNA tRNA genes has not been studied in human populations, nor have the significances of the various polymorphisms been determined in terms of the energetic demands of tRNA when folding into the secondary structure. Furthermore, the effect of selection on sequence variation in tRNA genes has not been examined in detail. Selection on genome regions should affect the respective allele frequency distributions, because polymorphisms under negative selection tend to be less frequent than neutral polymorphisms, whereas the opposite is true for polymorphisms that are subject to positive selection. These data would be of great importance in the search for possible pathogenic mutations and mildly deleterious polymorphisms in mtDNA. We therefore determined the sequences of the mtDNA tRNA genes in 477 Finns, obtained 435 other European sequences (Herrnstadt et al. 2002), and examined the variation in these genes. Furthermore, we analyzed the energetic consequences of the 96 polymorphisms found in these two data sets or reported in the Mitomap database.
Subjects and Methods
Subjects and Samples
A total of 477 blood samples from persons who reported that they themselves and their mothers were free of diabetes mellitus, sensorineural hearing impairment, and neurological ailments were obtained at Finnish Red Cross offices in central and northern Finland (Finnilä et al. 1999; Meinilä et al. 2001). After obtaining informed consent, the samples were rendered anonymous. The study was approved by the Ethics Committee of the Medical Faculty, University of Oulu, and the Ethics Committee of the Finnish Red Cross.
In addition, 435 European tRNA-encoding sequences (Herrnstadt et al. 2002) from population controls or patients with neurodegenerative disease in the U.K. and U.S.A. were downloaded from the Mitokor Web site (http://www.mitokor.com/science/560mtdnasrevision.php) in May 2003.
Molecular Methods
PCR for CSGE.
Total DNA was isolated from the blood cells using the QIAamp Blood Kit (Qiagen, Hilden, Germany), and the mitochondrial tRNA genes were analyzed by conformation-sensitive gel electrophoresis (CSGE). Thirteen pairs of primers were designed for amplification of the mtDNA fragments (size range, 261 bp to 428 bp) that harbored the 22 tRNA genes (Table 4). It is possible by CSGE to detect substitutions that are 44–50 nucleotides from the nearest end of the fragment (Ganguly et al. 1993; Finnilä et al. 2000). To confirm that we had not failed to detect any substitutions, primers were designed to amplify at least 56 extra nucleotides flanking the tRNA genes.
The template DNA was amplified in a total volume of 50 μl by PCR in 30 cycles of denaturation at 94°C for 1 min, annealing at a temperature specific to the primer for 1 min, and extension at 72°C for 1 min, with a final extension at 72°C for 10 min. The quality of the amplified fragment was estimated visually on a 1.5% agarose gel, after which a suitable amount of the PCR product, usually 2–8 μl, was taken for heteroduplex formation. This was allowed to occur autogenously, and each amplified fragment was mixed with the corresponding fragment amplified on a control template with a known sequence (Finnilä et al. 2000). The amplified fragments were denatured at 95°C for 5 min and the heteroduplexes were subsequently allowed to anneal at 68°C for 30 min.
CSGE was carried out as described previously (Körkkö et al. 1998; Finnilä et al. 2000). The heteroduplex samples were electrophoresed through a polyacrylamide gel at a constant voltage of 400 V overnight at room temperature. The bands in the gel were stained with ethidium bromide and visualized under UV light.
Sequencing.
Differences in the mobility of PCR fragments in CSGE were analyzed by automated sequencing (ABI PRISM 377 Sequencer using the DYEnamic ET terminator Cycle Sequencing Kit, Amersham Pharmacia Biotech Inc., Piscataway, NJ) after treatment with exonuclease I and shrimp alkaline phosphatase (Werle et al. 1994). The primers used for sequencing were the same as those used in the amplification reactions for CSGE. Polymorphisms are reported relative to the revised Cambridge Reference Sequence (rCRS) (Anderson et al. 1981; Andrews et al. 1999).
Statistical Analysis
Sequence data on the mtDNA tRNAs were used to calculate molecular diversity indices within populations by means of the ARLEQUIN 2.0 software package (Schneider et al. 2000). An analysis of molecular variance (AMOVA) was used to detect variance among and within populations.
The paired-samples t test was used to detect differences between the wild-type tRNA minimum free energies and those of the variant tRNAs, and the Kolmogorov–Smirnov test was used to examine the distributions of variables for normality.
Selection on genome regions should affect the respective allele frequency distributions. Absence of recombination is a reasonable assumption for mtDNA (Kraytsberg et al. 2004; Piganeau and Eyre-Walker 2004), and therefore, it is possible to compare the allele frequency distributions between mtDNA regions by using simple nonparametric rank order tests when the comparisons are being made in the same samples. If allele frequencies are lower in one region than another, there is selection on at least one of the two regions. Allele frequency distributions were compared between genome regions by using nonparametric rank order tests as implemented in R 1.8.1 (Ihaka and Gentleman 1996).
Determination of tRNA Structures
The consensus secondary structures of the mitochondrial tRNA molecules were first visualized using databank information available from URL http://www.uni-bayreuth.de/departments/ biochemie/sprinzl/trna/index.html/ (Sprinzl et al. 1998). The minimum free energy (ΔG) was calculated for all positions that harboured a polymorphism, using the latest free energy rules as implemented in mfold version 3.1 (available from URL http://www.bioinfo.rpi.edu/applications/mfold/old/rna/ [Zuker et al. 1999; Mathews et al. 1999]). Default settings were used for the folding temperature (37°C) and ionic conditions (1 M NaCl; no divalent ions). The percent suboptimality number was 5 and the upper bound on the number of computed foldings was 50. The maximum distance between paired bases was unlimited. The calculations were first carried out for the wild-type sequences. The constraint options of the program were used to force the stems to a consensus structure in which the amino acid acceptor stem was 7 bp, the D stem 4 bp, the anticodon stem 5 bp, and the T stem 5 bp (Helm et al. 2000). The anticodon loop was forced to contain 7 nt. In the case of 12 tRNA sequences (LeuUUR, Ile, Gln, Ala, Cys, SerUCN, Asp, His, SerAGY, LeuCUN, Thr, Pro) the algorithm suggested base pairing in a loop, which was then prevented by using the constraint options. Base pairing in a loop was accepted only in cases where a polymorphism created an extension of the stem. Unfolding of a stem was always accepted.
Results
Sequence Analysis of mt-tRNA Genes in 477 Finns
We determined the nucleotide sequences of the 22 tRNA genes in the mtDNA of 477 Finns. CSGE was used to detect differences in the mobility of amplified fragments, and the fragments showing differential mobility were then sequenced. Sequence information was thus obtained for 717,408 nucleotide positions. We found 27 polymorphic sites in the tRNA genes, with a total frequency of 314 polymorphisms among the 477 subjects, giving frequencies of 0.66 polymorphisms per subject and 0.438 × 10−3 per nucleotide site (Table 1). Twelve of the 27 polymorphic sites were located in the stems and 15 in the loops or other non-base-paired domains (Fig. 1a). All the polymorphisms were transitions, and purine and pyrimidine bases were equally affected, while thymidine and adenine nucleotides were affected approximately twice as often as cytosine and guanine nucleotides. A heteroplasmic transition 14696A>G was identified, whereas all the remaining polymorphisms were homoplasmic. The highest number of polymorphic sites was found in tRNA genes for Thr (five polymorphic sites), Met (three polymorphic sites), Cys (three polymorphic sites), and His (three polymorphic sites), whereas no polymorphic sites were detected in eight tRNA genes (Phe, Val, LeuUUR, Ile, Asn, Tyr, Lys, Pro). Haplogroup-specific polymorphisms were found at positions 10034 (I), 10463 (T), 12308 (KU), 15904 (V), 15924 (I), and 15928 (T). Two polymorphisms, 10463T>C and 15924A>G, were found to be homoplasic, i.e., they had occurred at least twice in evolution. Haplotypes of the samples were then constructed on the basis of the tRNA sequences. The most common haplotype was the one that was identical to the rCRS, and the samples representing haplogroups H, J, W, X, and Z belonged to this haplotype.

Secondary structure of human mitochondrial tRNA showing distribution of polymorphic sites among (a) 477 Finnish sequences and (b) 435 European sequences. 12311insA, 3′ to position 48, is not shown. The nucleotide positions are coded according to the number of different polymorphisms that hit the site, and the legend in the inset shows the frequencies, con 1, connector 1; con 2, connector 2.
Comparison of tRNA Sequences Between the Finns and Other Europeans
Complete mtDNA coding region sequences have been reported for 435 Europeans in British or U.S. populations (Herrnstadt et al. 2002). There were 52 polymorphic sites in the 22 tRNA genes including 49 transitions, 2 transversions, and 1 insertion (Fig. 1b). In total, there were 331 polymorphisms among the 435 subjects, giving frequencies of 0.76 polymorphisms per subject and 0.506 × 10−3 per nucleotide site (Table 2). Each tRNA molecule except that for proline harbored at least one polymorphic site, and the highest number of sites was found in the tRNA gene for threonine with eight polymorphic sites. Eight polymorphisms were homoplasic (positions 593, 5773, 7581, 10410, 10463, 15924, 15928, and 15930).
The standard indices of genetic heterogeneity were quite similar in the Finns and the Europeans (Table 3). The most common haplotype in the two groups was one belonging to haplogroup H, with a frequency of 42% among the Finns and 51% among the Europeans, and the second most common belonged to haplogroup U and had a frequency of 30% among the Finns and 13% among the Europeans. In total, 13 haplotypes were common to the Finns and the Europeans, and some 87% of the samples in both populations belonged to these haplotypes. The frequency of private haplotypes was fairly low within the two populations (Table 3), and a total of 50 haplotypes were private in the populations. AMOVA suggested that 97.5% of the variance was due to within-population variation and 2.5% was due to variation among populations.
Analysis of Allele Frequency Distributions in tRNA Genes
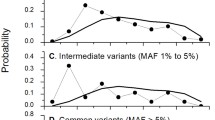
Comparisons of allele frequency distributions revealed significant differences between tRNA domains (Fig. 3A) and between synonymous sites and tRNA domains (Fig. 3B). Allele frequency distributions differed between the nine tRNA domains among 912 tRNA sequences (p = 2.7 × 10−5) (Fig. 3A). T and D stems were more conserved than their loop counterparts (T stem vs. TψC loop, p = 0.0048; D stem vs. DHU loop, p = 0.021). The distributions differed between the four loops (p = 0.033) but not between the four stems (p = 0.71). The anticodon loop was more conserved than the variable loop (p = 0.0014). The proportion of segregating sites was lowest in the anticodon stem, but the frequencies of the derived alleles in these sites were higher in the anticodon stem than in other tRNA domains (anticodon stem vs. variable loop, p = 0.012; anticodon stem vs. TψC loop, p = 0.014; anticodon stem vs. DHU loop, p = 0.0047; anticodon stem vs. amino acid stem, p = 0.017; anticodon stem vs. anticodon loop, p = 0.0090; anticodon stem vs. connectors, p = 0.079; anticodon stem vs. D stem, p = 0.027; anticodon stem vs. T stem, p = 0.0090).
The allele frequencies within the various tRNA domains and those at synonymous and nonsynonymous sites were examined in the 627 sequences that included the coding region (Fig. 3B). In comparison to synonymous sites, the allele frequencies were lower in nonsynonymous sites (p < 2 × 10−16) and in each of the tRNA domains except the variable loop (synonymous vs. variable loop, p = 0.22; synonymous vs. TψC loop, p = 0.0031; synonymous vs. DHU loop, p = 0.0041; synonymous vs. amino acid stem, p = 3.0 × 10−9; synonymous vs. anticodon loop, p = 2.2 × 10−5; synonymous vs. connectors, p = 0.0032; synonymous vs. T stem, p = 2.5 × 10−8; synonymous vs. D stem, p = 2.3 × 10−6; synonymous vs. anticodon stem, p = 3.1 × 10−8). Differences between nonsynonymous sites and tRNA domains were not significant, except for the variable loop (p = 0.00026). The allele frequencies at the segregating sites in the anticodon stem were higher than those at the synonymous sites (p = 0.0012).
tRNA Structure
We calculated the free energies for the wild-type tRNA molecules (ΔG(wt)) and for the 96 polymorphic tRNA molecules (ΔG(pmf)) that were detected among the 477 Finns or in the 435 European sequences in the MitoKor dataset or had been reported as mtDNA coding region sequence polymorphisms in the Mitomap (URL: http://mitomap.org). The calculations were performed using the latest free energy rules as implemented in mfold version 3.1 (Zuker et al. 1999; Mathews et al. 1999). The algorithm did not allow single base pairs in the stem, and consequently 4 of the wild-type tRNAs and 10 tRNAs harboring a polymorphism differed in their secondary structures from those suggested previously (Sprinzl et al. 1998) even after the application of constraints that forced a double-helical structure in the stems and an unpaired structure in the loops.
The minimum free energy was identical in 47 pairs of wild-type tRNA and polymorphic tRNA, more negative in the polymorphic tRNA in 26 pairs, and less negative in the polymorphic tRNA in 23 pairs. The minimum free energies were normally distributed among the 22 wild-type tRNAs, whereas those of the 96 polymorphic tRNAs departed from a normal distribution (p = 0.008, Kolmogorov–Smirnov test). The minimum free energies were then estimated separately for each domain of the tRNAs, and the difference in ΔG(wt) − ΔG(pmf) was calculated. The four helices were found to differ from each other (p = 0.003) in ΔG(wt) − ΔG(pmf). The mean difference was small in the amino acid acceptor stem, −0.085 ± 0.559, and in the dihydrouridine stem, −0.015 ± 0.492, but it was markedly larger in the T stem, 0.209 ± 1.036, and in the anticodon stem, −0.141 ± 0.564. Pairwise comparison of the minimum free energy between the helical domain of the wild-type molecule and that of the polymorphic molecule suggested that the differences in the anticodon stem (p = 0.015; paired-samples t test) and T stem (p = 0.05) were significant. Seven polymorphic positions were found in the anticodon stem and six in the T stem. Interestingly, the consensus base pairs 29·41 and 30·40 in the anticodon stem and base pair 50·64 in the T stem were devoid of polymorphisms (Fig. 1).
The difference in ΔG(wt) − ΔG(pmf) was then plotted against the frequency of the polymorphism among the Finns (Fig. 2), whereupon it emerged that the largest differences in ΔG(wt) − ΔG(pmf) tended to be in the polymorphisms that were present at a low frequency in the population. A cutoff frequency of 0.01 was used to stratify the 26 homoplasmic Finnish polymorphisms into two groups. The difference in ΔG(wt) − ΔG(pmf) was −0.3100 ± 0.9556 (mean ± SD) among the polymorphisms with a frequency <0.01 and −0.2411 ± 1.0812 among those with a frequency ≥0.01.

The difference ΔG(wt) − ΔG(pmf) as a function of the frequency of the polymorphism in the Finnish population. Black square, seven polymorphisms with ΔG(wt) equal to ΔG(pmf) and with a population frequency of 0.21%.

Comparisons of allele frequency distributions in the various domains of tRNA genes and synonymous and nonsynonymous sites in protein-coding genes. A tRNA domains in 912 tRNA sequences. B tRNA domains and synonymous and nonsynonymous sites in 627 sequences that included the coding region. Horizontal bars indicate the relative frequencies of mtDNA sites having the depicted absolute frequency of the derived allele (shading) among each set of sequences. The total number of sites belonging to each domain is shown at the right. Derived alleles were identified as differences from an African outgroup (GenBank accession number AF347015). Invariant sites that differed from the outgroup were excluded from the analyses. tRNA domains were defined according to Helm et al. (2000). Synonymous (fourfold degenerate) and nonsynonymous (nondegenerate) sites were defined according to AF347015. Initiation and termination codons were excluded. Allele frequency distributions were compared using Wilcoxon rank sum test for comparisons between two domains and Kruskal–Wallis rank sum test for comparisons among several domains.
Discussion
We obtained sequence information on more than 1.3 Mb of the 22 mtDNA-encoded tRNA genes in 477 Finns and 435 other Europeans (Herrnstadt et al. 2002). The tRNA molecules showed fairly low sequence variation by comparison with the structural genes, ribosomal RNA genes, and control region (Moilanen et al. 2003), their mean diversity being only 70% of that in the hypervariable segment I (HVS-I) among Europeans and 75% of that among the Finns. In keeping with this low diversity, the two most common haplotypes encompassed two-thirds of all the tRNA sequences, and the proportion of private haplotypes was quite low. AMOVA showed that 2.5% of the total variation in the sample was due to variation among the two populations. Actual comparison of the two populations may not be feasible, however, because the frequencies of mtDNA haplogroups differed between the samples (p < 0.001; exact test of population differentiation).
An interspecies comparison has revealed that mitochondrial tRNAs harbor invariant positions less frequently than do nuclear tRNAs (Lynch 1996). We found that the human mitochondrial tRNAs were variably subject to polymorphisms, as threonine tRNA harbored eight polymorphic sites, while the tRNAs for Val, Phe, LeuUUR, Tyr, and SerUCN harbored only one polymorphic site each among these 912 Europeans. Interestingly, the Mitomap list of disease-causing mutations in mtDNA-encoded tRNA genes gives a quite different distribution, four or more disease-causing mutation sites being present in only 6 (Phe, LeuUUR, Ile, SerUCN, Lys and Gly) of the 22 tRNA genes (27%), with 3 of these 6 belonging to those genes that rarely harbor polymorphisms. When the tRNAs were grouped according to the number of polymorphic sites, those tRNAs with only one or two polymorphisms harbored on average 5.7 ± 5.8 sites with a disease-causing mutation and those with more than two polymorphisms an average of 1.7 ± 1.1 such sites (p = 0.047 for the difference between the groups; t test). This relationship was most apparent in MTTL1, which has been considered to be a mutational hot spot (Moraes et al. 1993). Mitomap lists 21 disease-causing mutations in MTTL1, but we found only one polymorphic site in this tRNA gene among 912 Europeans. Thus there appears to be an inverse relationship between the number of deleterious mutation sites and the number of polymorphic sites in the tRNA, suggesting that variation in some tRNAs may be more deleterious than that in others.
Generally, the tRNA genes have been shown to be more conserved than synonymous sites (Pesole et al. 1999; Moilanen et al. 2003). We found here that different selective constraints are apparently imposed on entire tRNA genes, and in addition, we found evidence for similar differences between various tRNA structural domains. Comparisons of allele frequency distributions revealed significant differences between tRNA domains and between synonymous sites and tRNA domains. The variable loop was the least conserved and the anticodon stem was the most conserved of tRNA domains, and all domains except the variable loop were more conserved than the synonymous sites in the protein-coding genes. Furthermore, the allele frequencies of segregating sites in the anticodon stem were higher than expected by comparison to other tRNA domains or synonymous sites, suggesting that the observed polymorphisms in the anticodon stem may be either neutral or subject to positive selection.
In order to examine the structure–function relationships, we used the mfold program to predict optimal secondary structures for the mtDNA- encoded tRNAs through free energy minimization (Zuker et al. 1999; Mathews et al. 1999), in which the size of the minimum free energy change indicates the “driving force” behind the reaction, and the more negative the ΔG is, the more likely is the process. We found wide spectrum of differences in the ΔG of the mitochondrial tRNAs. The largest negative differences in ΔG(wt) −ΔG(pmf) < −3 were found in the polymorphisms 10001T>C, 10014G>A, 15927 G>A, and 15928G>A, suggesting that the polymorphic variant was energetically less favorable than the wild-type variant, while the largest positive differences (ΔG(wt) − ΔG(pmf) > 3), suggesting that the polymorphic variant was energetically more favorable than the wild-type variant, were found in the polymorphisms 8343A>G, 5634A>G, 4318C>G, 14696A>G, and 4310A>G. Polymorphisms differing greatly from the wild type are nonneutral.
The distribution of the minimum free energies of the polymorphic tRNAs deviated from normality, suggesting negative selection against some of the tRNA variants. The largest differences in minimum free energies were found between the wild-type tRNAs and the variant tRNAs in the anticodon stem. A reduction in the binding stability of the anticodon stem of the variant tRNAs would result in a less favorable conformation and would suggest a basis for negative selection. Interestingly, we found that the largest differences in ΔG(wt) − ΔG(pmf) tended to be in the polymorphisms that were present at a low frequency in the Finnish population. Such a frequency pattern would infer that the energy penalty for polymorphisms may be high and that some polymorphisms are faced with negative selection. This suggestion is supported by a recent analysis on 68 pathogenic mutations and 64 polymorphisms, which has shown that sites harboring polymorphisms tolerate more variability than those harboring pathogenic mutations (Florentz and Sissler 2001).
A recent survey of variation in 840 sequences on nonsynonymous variation revealed that nonconservative changes are more likely among private mutations and among nonhomoplasic mutations, suggesting that mtDNA variation in structural genes is non-neutral (Moilanen and Majamaa 2003). It is more difficult to assess the basis for negative selection in the case of tRNA molecules than in the case of the genes encoding subunits of the respiratory chain. Negative selection may be based on changes in the overall stability of the tRNA, on the extent or quality of the posttranscriptional modifications, or on interactions with aminoacyl tRNA synthetases, translation factors or ribosomal components (Söll and RajBhandary 1995).
Approximately 6% of the nucleotides in animal mitochondrial tRNAs are modified posttranscriptionally (Brulé et al. 1998) and the loops are most often the sites of modifications (Agris 1996). A structural role for such modified nucleotides is suggested by the fact that they are more often conserved than not. Furthermore, leucine tRNAs harboring the deleterious 3243A>G mutation and lysine tRNAs harboring the deleterious 8344A>G mutation have been shown to differ in the quality and extent of their posttranscriptional modifications (Helm et al. 1999). However, apart from the qualitative changes in the tRNA molecule, the consequences of posttranscriptional modification are unclear, although they seem to enhance stability of tRNAs or enhance conformational dynamics (Agris 1996). A change in tRNA modification could lead to a decrease in tRNA affinity for the respective aminoacyl synthetase (Muramatsu et al. 1988; Perret et al. 1990). Furthermore, posttranscriptional modifications may alter the specificity or stability of the tRNA or change its affinity for its codon. Whatever the consequence may be, the expected net effect would be a decrease in mitochondrial protein synthesis, which has been verified for many pathogenic mtDNA mutations (Rossignol et al. 2003).
Mature tRNA molecules interact with many proteins including aminoacyl tRNA synthetases, translation factors, and ribosomal components (Söll and RajBhandary 1995), and such proteins may be more or less tolerable for structural changes or instability of mutated tRNA molecules. However, it is unlikely that all tRNA variants that we observed are equally well tolerated by their partner proteins, because in that case variants with large ΔG and those with small ΔG should be found at similar frequencies. Mutations could also affect tRNA function by removing a nucleotide that is important in recognition by a partner protein, such as aminoacyl tRNA synthetase (Giegé et al. 1998). Mutations involving such binding sites may not necessarily cause significant changes in the structure of the tRNA molecule, and therefore it is likely that some mutations with small ΔG may also be subject to negative selection. The assumption that the pathogenic potential of a tRNA mutation is not determined solely by structural alterations is also supported by the finding that structural effects of clearly pathogenic mutations are not necessarily dramatic (Florentz and Sissler 2001).
We found that variation in the tRNA genes was low in European mtDNAs and the variants were unevenly distributed among the 22 tRNA genes, as the threonine tRNA harbored eight polymorphic sites, whereas five tRNAs harbored only one polymorphic site each. Calculation of the minimum free energies provided a method to evaluate the structural consequences of the various polymorphic variants. The ΔG’s of the polymorphic tRNAs varied considerably, but the difference in ΔG between the wild type and the polymorphic variant was small in the majority of tRNAs, suggesting that these variants are neutral. Interestingly, largest differences in ΔG’s tended to occur in those polymorphic tRNAs that were present at a low frequency in the Finnish population, suggesting that such variants are under negative selection. Changes in posttranscriptional modifications of tRNAs or changes in interactions of tRNAs with certain partner proteins may also contribute to such a selection, however. These results extend our recent data (Moilanen and Majamaa 2003) and suggest that some polymorphisms in the tRNA genes are not neutral and may have a mildly deleterious potential. Minimum free energy calculations in large sequence sets may provide a means for evaluating the pathogenic potential of polymorphisms in tRNAs, but these calculations do not reveal any simple basic features for predicting the pathogenic nature of tRNA variants.
References
PF Agris (1996) ArticleTitleThe importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function Prog Nucleic Acids Res Mol Biol 53 79–129 Occurrence Handle1:CAS:528:DyaK28XktlCmsrY%3D
S Anderson AT Bankier BG Barrel MHL De Bruijn AR Coulson J Drouin IC Eperon F Sanger PH Schreier AJH Smith R Staden IG Young (1981) ArticleTitleSequence and organization of human mitochondrial genome Nature 290 457–465 Occurrence Handle10.1038/290457a0 Occurrence Handle1:CAS:528:DyaL3MXlt1OlsL8%3D Occurrence Handle7219534
RM Andrews I Kubacka PF Chinnery RN Lightowlers DM Turnbull N Howell (1999) ArticleTitleReanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA Nat Genet 23 147 Occurrence Handle10.1038/13779 Occurrence Handle1:CAS:528:DyaK1MXmtlOhtLY%3D Occurrence Handle10508508
H Brulé WM Holmes G Keith R Giegé C Florentz (1998) ArticleTitleEffect of a mutation in the anticodon of human mitochondrial tRNAPro on its post-transcriptional modification pattern Nucleic Acids Res 26 536–543
S Finnilä IE Hassinen K Majamaa (1999) ArticleTitleRestriction fragment analysis as a source of error in detection of heteroplasmic mtDNA mutations Mutat Res Genomics 406 109–114
S Finnilä IE Hassinen L Ala-Kokko K Majamaa (2000) ArticleTitlePhylogenetic network of the mtDNA haplogroup U in northern Finland based on sequence analysis of the complete coding region by conformation-sensitive gel electrophoresis Am J Hum Genet 66 1017–1026 Occurrence Handle10712215
C Florentz M Sissler (2001) ArticleTitleDisease-related versus polymorphic mutations in human mitochondrial tRNAs EMBO Rep 21 481–486
AJ Fry (1999) ArticleTitleMildly deleterious mutations in avian mitochondrial DNA: evidence from neutrality tests Evolution 53 1617–1620 Occurrence Handle1:CAS:528:DyaK1MXnslGhsLY%3D
A Ganguly MJ Rock DJ Prockop (1993) ArticleTitleConformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes Proc Natl Acad Sci USA 90 10325–10329 Occurrence Handle1:CAS:528:DyaK2cXhs1Ch Occurrence Handle8234293
R Giege M Sissler C Florentz (1998) ArticleTitleUniversal rules and idiosyncratic features in tRNA identity Nucleic Acids Res 26 5017–5035 Occurrence Handle1:CAS:528:DyaK1cXnvF2jtLY%3D Occurrence Handle9801296
A Helgason S Sigurdardottir JR Gulcher R Ward K Stefansson (2000) ArticleTitleMtDNA and the origin of the Islanders: Deciphering signals of recent population history Am J Hum Genet 66 999–1016 Occurrence Handle1:CAS:528:DC%2BD3cXisFShsbc%3D Occurrence Handle10712214
M Helm C Florentz A Chomyn G Attardi (1999) ArticleTitleSearch for differences in post-transcriptional modification patterns of mitochondrial DNA-encoded wild-type and mutant human tRNALys and tRNALeu(UUR) Nucleic Acids Res 27 756–763 Occurrence Handle1:CAS:528:DyaK1MXhtFGms74%3D Occurrence Handle9889270
M Helm H Brulé D Friede R Giege D Putz C Florentz (2000) ArticleTitleSearch for characteristic structural features of mammalian mitochondrial tRNAs RNA 6 1356–1379 Occurrence Handle1:CAS:528:DC%2BD3cXnvF2ltbY%3D Occurrence Handle11073213
C Herrnstadt JL Elson E Fahy G Preston DM Turnbull C Anderson SS Ghosh JM Olefsky MF Beal RE Davis N Howell (2002) ArticleTitleReduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups Am J Hum Genet 70 1152–1171 Occurrence Handle10.1086/339933 Occurrence Handle1:CAS:528:DC%2BD38XjsFGns74%3D Occurrence Handle11938495
R Ihaka R Gentleman (1996) ArticleTitleR: a language for data analysis and graphics J Comp Graph Stat 5 299–314
RA Kittles AW Bergen M Urbanek M Virkkunen M Linnoila D Goldman JC Long (1999) ArticleTitleAutosomal, mitochondrial, and Y chromosome DNA variation in Finland: Evidence for a male-specific bottleneck Am J Phys Anthropol 108 381–399 Occurrence Handle1:STN:280:DyaK1M3ksVeqsg%3D%3D Occurrence Handle10229384
J Körkkö S Annunen T Pihlajamaa DJ Prockop L Ala-Kokko (1998) ArticleTitleConformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturating gradient gel electrophoresis and nucleotide sequencing Proc Natl Acad Sci USA 95 1681–1685
Y Kraytsberg M Schwartz TA Brown K Ebralidse WS Kunz DA Clayton J Vissing K Khrapko (2004) ArticleTitleRecombination of human mitochondrial DNA Science 304 981 Occurrence Handle10.1126/science.1096342 Occurrence Handle1:CAS:528:DC%2BD2cXktV2iu7Y%3D Occurrence Handle15143273
M Lynch (1996) ArticleTitleMutation accumulation in transfer RNA:s: molecular evidence for Muller’s ratchet in mitochondrial genomes Mol Biol Evol 13 209–220 Occurrence Handle1:CAS:528:DyaK28XhtVylu7w%3D Occurrence Handle8583893
DH Mathews J Sabina M Zuker DH Turner (1999) ArticleTitleExpanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure J Mol Biol 288 911–940 Occurrence Handle1:CAS:528:DyaK1MXjtVers7Y%3D Occurrence Handle10329189
M Meinilä S Finnilä K Majamaa (2001) ArticleTitleEvidence for mtDNA admixture between the Finns and the Saami Hum Hered 52 160–170 Occurrence Handle11588400
JS Moilanen S Finnilä K Majamaa (2003) ArticleTitleLineage-specific selection in human mtdna: lack of polymorphisms in a segment of MTND5 gene in haplogroup Mol Biol Evol 20 2132–2142 Occurrence Handle1:CAS:528:DC%2BD2cXnt12nsQ%3D%3D Occurrence Handle12949126
JS Moilanen K Majamaa (2003) ArticleTitlePhylogenetic network and physicochemical properties of nonsynonymous mutations in the protein-coding genes of human mitochondrial DNA Mol Biol Evol 20 1195–1210 Occurrence Handle1:CAS:528:DC%2BD3sXms1Wks78%3D Occurrence Handle12777521
CT Moraes F Ciacci E Bonilla C Jansen M Hirano N Rao RE Lovelace LP Rowland EA Schon S DiMauro (1993) ArticleTitleTwo novel pathogenic mitochondrial DNA mutations affecting organelle number and protein synthesis Is the tRNALeu(UUR) gene an etiologic hot spot? J Clin Invest 92 2906–2915 Occurrence Handle1:CAS:528:DyaK2cXltFyjsQ%3D%3D
T Muramatsu K Nishikawa F Nemoto Y Kuchino S Nishimura T Miyazawa S Yokoyama (1988) ArticleTitleCodon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification Nature 336 179–181 Occurrence Handle1:CAS:528:DyaL1MXjsFCqtQ%3D%3D Occurrence Handle3054566
N Neckelmann K Li RP Wade R Shuster DC Wallace (1987) ArticleTitlecDNA sequence of a human skeletal muscle ADP/ATP translocator: lack of a leader peptide, divergence from a fibroblast translocator cDNA, and coevolution with mitochondrial DNA genes Proc Natl Acad Sci USA 84 7580–7584 Occurrence Handle1:CAS:528:DyaL1cXhtVOhurw%3D Occurrence Handle2823266
V Perret A Garcia H Grosjean JP Ebel C Florentz R Giege (1990) ArticleTitleRelaxation of a transfer RNA specificity by removal of modified nucleotides Nature 344 787–789 Occurrence Handle1:CAS:528:DyaK3cXktlKqsrk%3D Occurrence Handle2330033
G Pesole C Gissi A De Chirino C Saccone (1999) ArticleTitleNucleotide substitution rate of mammalian mitochondrial genomes J Mol Evol 48 427–434 Occurrence Handle1:CAS:528:DyaK1MXhvFyqu7w%3D Occurrence Handle10079281
R Piercy KM Sullivan N Benson P Gill (1993) ArticleTitleThe application of mitochondrial DNA typing to the study of white Caucasian genetic identification Int J Legal Med 106 85–90 Occurrence Handle1:STN:280:ByuD3s7oslI%3D Occurrence Handle8217870
G Piganeau A Eyre-Walker (2004) ArticleTitleA reanalysis of the indirect evidence for recombination in human mitochondrial DNA Heredity 92 282–288 Occurrence Handle1:CAS:528:DC%2BD2cXisVWlu7Y%3D Occurrence Handle14747833
I Pult A Sajantila J Simanainen O Georgiev W Schaffner S Pääbo (1994) ArticleTitleMitochondrial DNA sequences from Switzerland reveal striking homogeneity of European populations Biol Chem Hoppe Seyler 375 837–840 Occurrence Handle1:CAS:528:DyaK2MXivF2jtL8%3D Occurrence Handle7710701
M Richards H Corte-Real P Forster V Macaulay H Wilkinson-Herbots A Demaine S Papiha R Hedges HJ Bandelt B Sykes (1996) ArticleTitlePaleolithic and neolithic lineages in the European mitochondrial gene pool Am J Hum Genet 59 185–203 Occurrence Handle1:CAS:528:DyaK28XksFSitr0%3D Occurrence Handle8659525
R Rossignol B Faustin C Rocher M Malgat JP Mazat T Letellier (2003) ArticleTitleMitochondrial threshold effects Biochem J 3 751–762
A Sajantila P Lahermo T Anttinen M Lukka P Sistonen ML Savontaus P Aula L Beckman L Tranebjaerg T Gedde-Dahl L Issel-Tarver A DiRienzo S Pääbo (1995) ArticleTitleGenes and languages in Europe: An analysis of mitochondrial lineages Genome Res 5 42–52 Occurrence Handle1:CAS:528:DyaK2MXotVeku7c%3D Occurrence Handle8717054
S Schneider D Roessli L Excoffier (2000) Arlequin ver. 2.000: A software for population genetics data analysis Genetics and Biometry Laboratory, University of Geneva Geneva, Switzerland
M Sprinzl C Horn M Brown A Ioudovitch S Steinberg (1998) ArticleTitleCompilation of tRNA sequences of tRNA genes Nucleic Acids Res 26 148–153 Occurrence Handle1:CAS:528:DyaK1cXovVansQ%3D%3D Occurrence Handle9399820
D Söll UL RajBhandary (1995) tRNA: Structure, biosynthesis, and function American Society for Microbiology Press Washington, DC
A Torroni K Huoponen P Francalacci M Petrozzi L Morelli R Scozzari D Obinu ML Savontaus DC Wallace (1996) ArticleTitleClassification of European mtDNAs from an analysis of three European populations Genetics 144 1835–1850 Occurrence Handle1:CAS:528:DyaK2sXkslymug%3D%3D Occurrence Handle8978068
DC Wallace (1995) ArticleTitleMitochondrial DNA variation in human evolution, degenerative disease, and aging Am J Hum Genet 57 201–223 Occurrence Handle1:CAS:528:DyaK2MXotFOjsbg%3D Occurrence Handle7668244
DC Wallace MD Brown MT Lott (1997) Mitochondrial genetics DL Rimoin JM Connor JRE Pyeritz AEH Emery (Eds) Emory and Rimoin’s principles and practise of medical genetics Churchill Livingstone London 277–332
E Werle C Schneider M Renner M Volker W Fiehn (1994) ArticleTitleConvenient single-step, one tube purification of PCR products for direct sequencing Nucleic Acids Res 22 4354–4355 Occurrence Handle1:CAS:528:DyaK2MXhvF2itbY%3D Occurrence Handle7937169
M Zuker DH Mathews DH Turner (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide J Barciszewski BFC Clark (Eds) RNA biochemistry and biotechnology Kluwer Academic Boston, MA 11–43
Acknowledgments
The expert technical assistance of Ms. Anja Heikkinen, Ms. Pirjo Keränen, and Ms. Irma Vuoti is acknowledged. This study was supported in part by grants from the Council for Health Research of the Academy of Finland (project 79843 to K.M. and 107490 to J.S.M.), the Sigrid Juselius Foundation, and the Medical Research Foundation of Oulu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Rüdiger Cerff
Rights and permissions
About this article
Cite this article
Vilmi, T., Moilanen, J.S., Finnilä, S. et al. Sequence Variation in the tRNA Genes of Human Mitochondrial DNA. J Mol Evol 60, 587–597 (2005). https://doi.org/10.1007/s00239-003-0202-1
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-0202-1