
Abstract
A gene complex, dot/icm, located in two independent chromosomal loci of L. pneumophila, the causative agent of Legionnaires' disease, is related to virulence. To investigate the evolutionary pattern of these pathogenicity islands of L. pneumophila, portions of four genes in the dot/icm complex, namely, dotA, dotB, icmB, and icmT, were amplified, sequenced, and phylogenetically analyzed, in addition to rpoB, which encodes an RNA polymerase β-subunit. The nucleotide sequences and phylogenetic analyses of these five genes of 96 L. pneumophila strains revealed that several subgroups of L. pneumophila proliferated clonally. However, incongruent gene tree topologies and the results of statistical testing (Templeton Willcoxon signed-ranked and incongruence length differences tests) indicated that the evolutionary histories of these genes within the pathogenicity islands are not uniform, and that they constitute a mosaic structure. In addition, the non-uniform grouping of some reference strains suggests that intraspecific recombination might be still occurring in nature or in the laboratory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The gram-negative respiratory pathogen Legionella pneumophila is a facultative intracellular bacterium that infects and grows within human alveolar macrophages and protozoan host cells (Winn 1999). It has been postulated that L. pneumophila forms pores in the macrophage membrane and enter a vacuole, thus avoiding fusion with lysosomes when it is taken up by macrophages (Swanson and Hammer 2000). It then replicates inside the alveolar macrophages causing a severe type of pneumonia called Legionnaires' disease (Vogel and Isberg 1999). That is to say, the most important characteristic of its virulence may be its ability to prevent phagosome-lysosome fusion (Vogel and Isberg 1999). The alteration of endocytic trafficking in phagosomes by L. pneumophila is mediated through the actions of products encoded by dot/icm (defect in organelle trafficking/intracellular multiplication) genes (Berger and Isberg 1993; Purcell and Shuman 1998; Setal et al. 1998; Vogel et al. 1998).
Twenty-four dot/icm genes are located in two unlinked 22-kb regions on the L. pneumophila chromosome (Segal et al. 1998; Vogel et al. 1998). Chromosomal region I contains the genes icmV, icmW, and icmX, and dotA, dotB, dotC, and dotD, and chromosomal region II the genes icmT, -S, -R, -Q, -P, -N, -M, -L, -K, -E, -G, -C, -D, -J, and -B, and tphA and icmF (Segal et al. 1998; Vogel et al. 1998; Vogel and Isberg 1999). The Dot/Icm complex is thought to constitute a type IV-like secretion system, which is capable of injecting effector protein into the host cell, that allows L. pneumophila to evade the endocytic pathways by modulating the activity of the host factors involved in vesicle trafficking (Nagai et al. 2002; Zink et al. 2002). Moreover, L. pneumophila mutants defective in the Dot/Icm transporter system cannot replicate intracellularly (Vogel et al. 1998; Andrews et al. 1998; Roy et al. 1998; Wiater et al. 1998; Mathews and Roy 2000), which implies that the dot/icm complex is indispensable for the intracellular survival of L. pneumophila, and that its mode of action is evolutionarily conserved (Hilbi et al. 2001).
It has been also reported that most of the Dot/Icm proteins share significant amino acid sequence similarity with those of plasmid-encoded conjugation systems, such as R64 (Segal and Shuman 1999; Komano et al. 2000). Thus, it has been suggested that the R64 transfer of L. pneumophila and L. pneumophila dot/icm systems have evolved from a common ancestral genetic system (Komano et al. 2000). In addition, the dotA gene has been detected in other Legionella species, such as L. micdadei, L. longbeachae, L. bozemanii, and L. gratiana (Nagai et al. 2002). In addition, homologues of icmT, icmS, and icmK have been found in Coxiella burnetii (Segal and Shuman 1999), which is the causative agent of Q-fever and which is closely related evolutionarily to Legionella (Weisburg et al. 1989). These relationships suggest that the dot/icm complex was transferred from a plasmid into an unknown common ancestor of Legionella and Coxiella, and has since evolved.
Thus, it is possible that the pathogenicity islands of the L. pneumophila, dot/icm complex, have evolved not only by plasmid incorporation into its chromosome, but also by the intraspecific recombination of complex after this plasmid incorporation. To verify this we analyzed the genetic structure of four genes (dotA, dotB, icmB, and icmT) in the dot/icm complex of L. pneumophila to investigate their evolutionary patterns. These four genes were selected on the basis of their importance in terms of their virulence and disparity from one another. Along with these genes, we also determined the sequence of a portion of the housekeeping gene, rpoB. This gene encodes the β-subunit of DNA-dependent RNA polymerase (Severinov et al. 1996), which has been suggested to be an alternative tool for determining the phylogeny of and for identifying several bacteria (Mollet et al. 1997; Kim et al. 1999; Ko et al. 2002). It was interesting to note that no concordance was found among the phylogenetic relationships inferred from the five gene sequences, suggesting the complex molecular evolution of the pathogenicity islands in L. pneumophila.
Materials and Methods
Bacterial Strains
Ninety-six strains of L. pneumophila were used for this analysis, including 17 reference strains. Reference strains in all serogroups (SGs) of L. pneumophila (SGs 1 to 15) were included in this study except SG 9, because of no amplification of dotB, icmB, and icmT. Four reference strains, ATCC 33152 (Philadelphia-1, type strain), ATCC 33153 (Knoxville-1), ATCC 43109 (OLDA), and SF9, belonged to SG 1. Of the 79 culture isolates, 76 strains were isolated from air-conditioner cooling water, and the three remaining strains (KP1, KP2, and KP3) were obtained from the lung tissue of pneumonia patients. These had been previously identified by serologic and biochemical testing. For rpoB, dotA, and icmB, all 96 strains were included in the nucleotide sequencing and analyses, but 37 strains, including 17 reference strains, were selected from each L. pneumophila subgroup (Ko et al. 2002) and analyzed for dotB and icmT.
Nucleotide Sequencing of Gene Fragments
The nucleotide sequences of the internal fragment of dotA, dotB, icmB, and icmT were determined along with rpoB. The primers used for this amplification and sequencing are shown in Table 1. For PCR, 20 pmol of each primer were added to a PCR mixture tube (AccuPower PCR PreMix; Bioneer, Daejeon, Korea), which contained 1 unit of Taq DNA polymerase, and each deoxynucleoside triphosphate at a concentration of 250 µM, 10 mM Tris-HCl (pH 8.3), 40 mM KCl, 1.5 mM MgCl2, and gel loading dye. The bacterial culture suspension (1 µl) was amplified directly by PCR. The final volume was adjusted to 20 µl with distilled water, and the reaction mixture was subjected to 30 cycles of amplification. Each cycle consisted of: 30 s at 95°C for denaturation, 30 s at 50–55°C for annealing, and 30 s at 72°C for extension, and this was followed by a final extension at 72°C for 5 min (model 9700 Thermocycler; Perkin-Elmer Cetus). PCR products were detected on 1.5% agarose gels stained with ethidium bromide and purified using a QIAEX II gel extraction kit (Qiagen, Hilden, Germany) for sequencing. The sequences of the purified PCR products were determined directly with forward and reverse primers using an ABI377 automated sequencer and a BigDye Terminator Cycle Sequencing kit (Perkin-Elmer Applied Biosystems, Warrington, United Kingdom). For the sequencing reaction, 30 ng of purified PCR products, 2.5 pmol of primer, and 4 µl of BigDye Terminator RR mix (Perkin-Elmer Applied Biosystems; part number 4303153) were mixed and adjusted to a final volume of 10 µl with distilled water. The reaction was run with 5% (vol/vol) dimethyl sulfoxide for 30 cycles of: 15 s at 95°C, 5 s at 50°C, and 4 min at 60°C.
Phylogenetic Analysis
The five individual gene data sets of the 17 reference strains were compared statistically for incongruence using the nonparametric Templeton Wilcoxon signed-rank (WS-R) test (O'Donnell et al. 2001) and the incongruence length differences (ILD) test (or the partition homogeneity test) (Farris et al. 1994; Cunningham 1997), both of which are implemented in the PAUP* package (Swofford 1999). The maximum-likelihood (ML) method was also used to determine the extent of congruence among the gene trees (Holmes et al. 1999; Feil et al. 2001). For each gene, differences in log likelihood (Δ-lnL) were calculated between the ML tree for a gene and the ML trees based on other genes. These differences in log likelihood were compared with those of 200 randomly generated trees for each gene. If the ML trees of each gene are congruent, then all gene trees should have smaller differences in log likelihood than those of the random trees (Holmes et al. 1999). This analysis was also performed using the PAUP* program (Swofford 1999).
The phylogenetic tree of each gene fragment sequence was constructed using the neighbor-joining method in PAUP*, with the ML distance option of the HKY85 substitution model and no among-site rate variation. All trees were rooted using the midpoint-rooting option. Branch supporting values were evaluated by performing 1000 bootstrap replications.
Nucleotide Sequence Accession Numbers
The nucleotide sequences determined in this study were submitted to the GenBank database. The accession numbers of the reference strains are AF367748, AF527122–AF527172, and AY036018–AY036052.
Results
Sequence Diversity
Sequences of rpoB, dotA, and icmB were obtained from 96 L. pneumophila strains and those of dotB and icmT from 37 strains. The unambiguously determined nucleotide sequences in this study ranged from 264 bp (icmT) to 360 bp (dotA) (Table 1). Sequences were edited and aligned using the EditSeq and MegAlign programs (Windows version 3.12e; DNASTAR, Madison, WI). No insertions or deletions whatsoever were observed in any regions sequenced during this study. At the nucleotide level, the maximum divergences of rpoB, dotA, dotB, icmB, and icmT were 12.7%, 21.9%, 9.6%, 9.0%, and 5.2%, respectively. The deduced amino acid sequences were compared with previously published sequences (Segal et al. 1998; Vogel et al. 1998). No amino acid substitution was found in RpoB. Polymorphisms were found at one site in DotB, six in IcmB, and five in IcmT, most of which were shown by two subspecies of L. pneumophila (data not shown). On the other hand, extensive substitutions, 48 out of 120 deduced amino acids, were present in DotA.
Incongruence of Sequence Data Sets
Both the Templeton WS-R test and the ILD test showed that the five individual gene data sets of the 17 reference strains should not be combined with each other. The Templeton WS-R test indicated that all data sets were significantly incongruent (p < 0.05). All the results of the ILD tests using 1000 replicates for 10 combined data sets also indicated low combinability (p < 0.01), i.e., incongruence (Cunningham 1997). The results of ML analysis of congruence between gene trees are presented in Table 2. The differences between the log likelihoods of trees (Δ-lnL) generated from the different genes fell inside the 99th percentile of the random tree topologies in two cases, the icmT tree in dotA and the icmB tree in icmT. Thus, all results showed that rpoB, dotA, dotB, icmB, and icmT of L. pneumophila might have been subjected to different evolutionary pathways.
Subgroups of L. pneumophila Strains
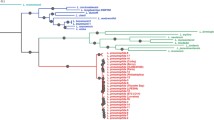
Gene trees inferred from the five gene fragments are shown in Figs. 1, 2, 3, 4, 5. Previously, the population of L. pneumophila strains was classified into six subgroups (P-I to -IV for L. pneumophila subsp. pneumophila, and F-I and -II for L. pneumophila subsp. fraseri), according to the rpoB and dotA gene analysis (Ko et al. 2002). Six subgroups were also valid in other gene (icmB, dotB, and icmT) trees with the exception of subgroup P-I in the icmB tree. Though subgroup P-III of subsp. pneumophila was close to the two subgroups of subsp. fraseri in the dotA tree (Fig. 2), two subspecies of L. pneumophila were not intermixed in the five gene trees indicating the genetic separation of the two subspecies (Ko et al. 2002). In the icmB tree, strains of the P-I subgroup did not form a monophyletic cluster. Instead, subgroup P-II was inserted within the main clade of subgroup P-I (Fig. 3), and the icmB sequences of subgroup P-II differed by only one or two nucleotides from those of subgroup P-I.

Neighbor-joining tree based on the rpoB gene sequence. Thirty-eight strains selected in dotB (Fig. 4) and in icmT (Fig. 5) are represented as asterisks at the right of the strain names. Designation of L. pneumophila subgroups, P-I to P-IV and F-I to F-II from Ko et al. (2002).



Neighbor-joining tree based on the dotB gene sequence.

Neighbor-joining tree based on the icmT gene sequence.
Interrelationship Between the L. pneumophila Subgroups
In contrast to the general conservation of subgroupings within L. pneumophila, interrelationships between the subgroups were not concordant (Figs. 1, 2, 3, 4, 5). No pair of gene trees showed the same tree topology among the subgroups. Though the two subgroups of L. pneumophila subsp. fraseri (F-I and F-II) were separated, they were nevertheless closely related with the rpoB, dotA, and dotB trees, and were not separated in the icmB and icmT trees. However, four subgroups of L. pneumophila subsp. pneumophila did not show consistent relationships, In the rpoB tree, which is believed to give reliable relationships (Ko et al. 2002), subgroups of L. pneumophila subsp. pneumophila formed a single cluster, which was distinctly separated from that of L. pneumophila subsp. fraseri (Fig. 1) and from the icmB and dotB trees.
Placements of Reference Strains of L. pneumophila in Gene Trees
Table 3 shows the subgroups of the 17 reference strains. Only the five reference strains of L. pneumophila subsp. pneumophila, ATCC 43109 (OLDA, SG 1), ATCC 33154 (SG 2), ATCC 33155 (SG 3), ATCC 33215 (SG 6), and ATCC 43290 (SG 12), and the two reference strains of L. pneumophila subsp. fraseri, ATCC 33156 (SG 4) and ATCC 33216 (SG 5), belonged to identical subgroups in all gene trees. The other reference strains clustered into different subgroups in different gene trees. For example, the Philadelphia-1 (ATCC 33152, SG 1) type strain of L. pneumophila belonged to P-I in the rpoB and dotA trees but to P-III in the icmB, dotB, and icmT trees. Morever, the dispositions of ATCC 33153 (Knoxville-1, SG 1), ATCC 43283 (SG 10), and ATCC 35251 (SG 15) into subgroups differed in each gene tree (Table 3). In addition, SF9 (SG 1), ATCC 33823 (SG 7), ATCC 35096 (SG 8), ATCC 43736 (SG 13), and ATCC 43703 (SG 14) did not belong to any subgroup in some of the gene trees, though they merged into one of the subgroups in others (Table 3), with the exception of ATCC 43130 (SG 11), which was not included in any subgroup in the five gene trees. All isolates consistently belonged to the same subgroups in the five gene trees without any exception, while several reference strains did not.
Discussion
The integration of foreign DNA into the bacterial chromosome is an important aspect of the evolution of genomes and the emergence of new pathogens (Hacker et al. 1997; Hacker and Kaper 2000). Genes encoding the proteins of the Type IV secretion system were introduced into bacterial genomes from plasmid, because their coding genes have been identified on many self-transmissible plasmids (Christie and Vogel 2000; Nagai and Roy 2001). The Dot/Icm system of L. pneumophila seems to be a protein transporter of the type IV secretion system, as is found in Brucella sp., Bordetella pertussis, Helicobacter pylori, and Rickettsia prowazekii (Christie and Vogel 2000). This study suggests that the dot/icm complex of L. pneumophila has evolved by the incorporation of a plasmid into its chromosome and by complex intraspecific recombination after this incorporation.
The gene trees inferred in this study show that genes within the dot/icm complex have experienced different evolutionary routes. If genes within the dot/icm complex had been transmitted en bloc and only vertically from one generation to the next, the phylogenetic relationships of each gene would be identical (Holmes et al. 1999; Kalia et al. 2002). However, the phylogenetic relationships among the six subgroups in the five gene trees were quite different (Figs. 1, 2, 3, 4, 5). Incongruence tests, such as the Templeton WS-R, the ILD, and the ML tests, confirmed the different evolutionary paths of the individual genes. Such incongruent tree topologies and sequence data sets can be explained by horizontal gene transfer (Feil and Spratt 2001; Feil et al. 2001).
However, the preservation of clonality in each subgroup, except subgroup P-I in the icmB tree, indicates that recombination among individuals of L. pneumophila is not as free as in H. pylori (Suerbaum et al. 1998). The congruence of subgroupings among the isolates may reflect genetic barriers to gene flow between the different subgroups within a population, i.e., cryptic speciation (Maynard Smith et al. 1993; van Belkum et al. 2001). Horizontal gene transfer in L. pneumophila may be sporadic and its limited clonality can be explained by the periodic emergence of fitter genotypes, which would give frequent rise to clones (Levin 1981). For example, the closeness of P-III, which is a subgroup of subsp. pneumophila, to L. pneumophila subsp. fraseri (F-I and F-II) in the dotA tree (Fig. 2) might be a relic of a past horizontal gene transfer of dotA from L. pneumophila subsp. fraseri to the ancestor of subgroup P-III (Ko et al. 2002). After the integration of foreign DNA, including dotA, the dotA gene of each subgroup might have evolved independently by mutation, and not by recombination. Thus, the clonality of each subgroup may have been preserved.
Strains of subgroup P-II were inserted into those of subgroup P-I, and two clades of subgroup P-I did not cluster with most isolates of subgroup P-I in the icmB tree (Fig. 3). This may indicate that horizontal gene transfer of icmB from subgroup P-I to subgroup P-II has occurred recently. Due to the relatively recent intragenic recombination of icmB, subgroups P-I and P-II are not separated clearly in a gene tree. After such a sporadic gene transfer, the icmB of subgroup P-II might have maintained segregated clonal proliferation from subgroup P-I by a single nonsynonymous mutation (R268 → K268). In addition, there is no evidence to suggest that the two distinct clades of subgroup P-I constitute other clonal complexes, because these isolates showed only one or two nucleotide differences and did not form a distinct cluster in any other gene tree.
On the other hand, intragenic recombination between certain subgroups of L. pneumophila subsp. pneumophila and subsp. fraseri seemed to have occurred long before or the mutation rate must have been high. Moreover, the intermediate positions of subgroup P-IV in the dotA tree (Fig. 2) and subgroup P-II in the icmT tree (Fig. 5), along with their preservation of clonality may support this proposal. Subgroup P-IV in icmB (Fig. 3) and subgroup P-II in dotB (Fig. 4) may also indicate the occurrence of sporadic recombination events or high mutation rates.
However, the inconsistency of subgroupings in several reference strains suggests that the recombination of genes within the dot/icm complex may be in progress. Whether the intragenic recombination of reference strains occurred naturally in the environment is not clear. Recently, it was reported that Lp01 and JR32, which originate from the Philadelphia-1 strain (SG 1, type strain), and which show genetic and phenotypic differences (Samrakandi et al. 2002), have been used in many molecular pathogenesis studies of L. pneumophila. Therefore, it was inferred that such differences are due to the different passage histories of strains. Coupled with such observations, it is feasible to suggest that such gene recombination in L. pneumophila may have occurred in the laboratory. It is important to note that such a recombination would affect the phenotype, i.e., the virulence and the susceptibility of a bacterium to antimicrobial agents (Feil and Spratt 2001; Samrakandi et al. 2002).
dotA showed more dissimilarity than the other four genes both in nucleotide and deduced amino acid sequences. This extensive diversity seems to have originated from its nature. DotA is a kind of polytopic membrane protein (Roy and Isberg 1997), and is secreted into culture supernatant by Dot/Icm transporter (Nagai and Roy 2001). Thus, the diversity of amino acids via lateral gene transfer and/or point mutations may increase the fitness of L. pneumophila in certain environmental niches, such as within a particular biofilm community or species of amoebae (Berger et al. 1994; Bumbaugh et al. 2002) in terms of immune surveillance system evasion. The high variation shown by dotA may also be related to adaptation and intracellular survival in different host species, such as various species of amoeba, ciliates, and other protozoans (Swanson and Hammer 2000). However, nucleotide and deduced amino acid divergences of the other genes within the pathogenicity islands (icmB, dotB, and icmT) were found to be similar to or lower than those of the housekeeping gene, rpoB. This indicates that though icmB, dotB, and icmT may have been exchanged by intraspecific recombination, no selective pressure has diversified them further.
This study suggests that complex recombination has occurred after the acquisition of blocks of pathogenicity islands in L. pneumophila. Such intraspecific recombination within pathogenicity islands has not been previously reported. In spite of a recent clonal proliferation and the presence of distinct subgroups in L. pneumophila, it is evident that there have been intraspecific recombinations and that the relationships between major subgroups of L. pneumophila should be depicted as a network rather than a tree. In other words, the pathogenicity islands of L. pneumophila are mosaic-like in structure.
References
HL Andrews JP Vogel RR Isberg (1998) ArticleTitleIdentification of linked Legionella pneumophila gene essential for intracellular growth and evasion of the endocytic pathway. Infect Immun 66 950–958 Occurrence Handle1:CAS:528:DyaK1cXhtlSktb8%3D Occurrence Handle9488381
KH Berger RR Isberg (1993) ArticleTitleTwo distinct defects in intracellular growth complement by a single genetic locus in Legionella pneumophila. Mol Microbiol 7 7–19 Occurrence Handle1:CAS:528:DyaK3sXns1yruw%3D%3D Occurrence Handle8382332
KH Berger JJ Merrian RR Isberg (1994) ArticleTitleAltered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol Microbiol 14 809–822 Occurrence Handle1:CAS:528:DyaK2MXivFansL0%3D Occurrence Handle7891566
AC Bumbaugh EA McGraw KI Page RK Selander TS Whittam (2002) ArticleTitleSequence polymorphism of dotA and mip alleles mediating invasion and intracellular replication of Legionella pneumophila. Curr Microbiol 44 314–322 Occurrence Handle10.1007/s00284-001-0024-6 Occurrence Handle1:CAS:528:DC%2BD38Xislaltbs%3D Occurrence Handle11927981
PJ Christie JP Vogel (2000) ArticleTitleBacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol 8 354–360 Occurrence Handle10.1016/S0966-842X(00)01792-3 Occurrence Handle1:STN:280:DC%2BD3cvptleltA%3D%3D Occurrence Handle10920394
CW Cunningham (1997) ArticleTitleCan three incongruence tests predict when data should be combined? Mol Biol Evol 14 733–740 Occurrence Handle1:CAS:528:DyaK2sXksVahs7c%3D Occurrence Handle9214746
JS Farris M Källersj AG Kluge C Bult (1994) ArticleTitleTesting significance of incongruence. Cladistics 10 315–319 Occurrence Handle10.1006/clad.1994.1021
EJ Feil EC Holmes DE Bessen M-S Chan NPJ Day MC Enright et al. (2001) ArticleTitleRecombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc Natl Acad Sci USA 98 182–187 Occurrence Handle10.1073/pnas.98.1.182 Occurrence Handle1:CAS:528:DC%2BD3MXkslOjsg%3D%3D Occurrence Handle11136255
EJ Feil BG Spratt (2001) ArticleTitleRecombination and the population structures of bacterial pathogens. Annu Rev Microbiol 55 561–590 Occurrence Handle10.1146/annurev.micro.55.1.561 Occurrence Handle1:CAS:528:DC%2BD3MXnslKjtLk%3D Occurrence Handle11544367
J Hacker JB Kaper (2000) ArticleTitlePathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54 641–679
J Hacker G Blum-Oehler I Mühldorfer H Tschäpe (1997) ArticleTitlePathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol 23 1089–1097 Occurrence Handle1:CAS:528:DyaK2sXit1ahtLo%3D Occurrence Handle9106201
H Hilbi G Segal HA Shuman (2001) ArticleTitleIcm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol 42 603–617 Occurrence Handle10.1046/j.1365-2958.2001.02645.x Occurrence Handle1:CAS:528:DC%2BD3MXos1Kgsb0%3D Occurrence Handle11722729
EC Holmes R Urwin MCJ Maiden (1999) ArticleTitleThe influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol 16 741–749 Occurrence Handle1:CAS:528:DyaK1MXjslGitb4%3D Occurrence Handle10368953
A Kalia BG Spratt MC Enright DE Besen (2002) ArticleTitleInfluence of recombination and niche separation on the population genetic structure of the pathogen Streptococcus pyogenes. Infect Immun 70 1971–1983 Occurrence Handle10.1128/IAI.70.4.1971-1983.2002 Occurrence Handle1:CAS:528:DC%2BD38Xitl2lsrw%3D Occurrence Handle11895961
B-J Kim S-H Lee M-A Lyu S-J Kim G-H Bai S-S Kim et al. (1999) ArticleTitleIdentification of mycobacterial species by comparative sequence analysis of the RNA polymerase gene (rpoB). J Clin Microbiol 37 1714–1720 Occurrence Handle1:CAS:528:DyaK1MXjslSrtL8%3D Occurrence Handle10325313
KS Ko HK Lee M-Y Park M-S Park K-H Lee S-Y Woo et al. (2002) ArticleTitlePopulation genetic structure of Legionella pneumophila inferred from RNA polymerase gene (rpoB) and DotA gene (dotA) sequences. J Bacteriol 184 2123–2130 Occurrence Handle10.1128/JB.184.8.2123-2130.2002 Occurrence Handle1:CAS:528:DC%2BD38Xis1Kqsbs%3D Occurrence Handle11914343
T Komano T Yoshida K Narahara N Furuya (2000) ArticleTitleThe transfer region of Incl1 plasmid R64: similarities between R64 tra and Legionella icm/dot genes. Mol Microbiol 35 1348–1359 Occurrence Handle10.1046/j.1365-2958.2000.01769.x Occurrence Handle1:CAS:528:DC%2BD3cXis1Wrsbc%3D Occurrence Handle10760136
BR Levin (1981) ArticleTitlePeriodic selection, infectious gene exchange, and the genetic structure of E. coli populations. Genetics 99 1–23 Occurrence Handle1:STN:280:Bi2C1cjot1w%3D Occurrence Handle7042453
J Maynard Smith NH Smith M O'Rourke BG Spratt (1993) ArticleTitleHow clonal are bacteria? Proc Natl Acad Sci USA 90 4384–4388 Occurrence Handle8506277
M Matthews CR Roy (2000) ArticleTitleIdentification and subcellular localization of the Legionella pneumophila IcmX protein: a factor essential for establishment of a replicative organelle in eukaryotic host cells. Infect Immun 68 3971–3982 Occurrence Handle10.1128/IAI.68.7.3971-3982.2000 Occurrence Handle1:CAS:528:DC%2BD3cXktlyntr0%3D Occurrence Handle10858211
CS Mintz (1999) ArticleTitleGene transfer in Legionella pneumophila. Microb Infect 1 1203–1209 Occurrence Handle10.1016/S1286-4579(99)00241-5 Occurrence Handle1:CAS:528:DC%2BD3cXlslaksQ%3D%3D
C Mollet M Drancourt D Raoult (1997) ArticleTitle rpoB sequence analysis as a novel basis of bacterial identification. Mol Microbiol 26 1005–1011 Occurrence Handle1:CAS:528:DyaK1cXls1E%3D Occurrence Handle9426137
H Nagai JC Kagan X Zhu RA Kahn CR Roy (2002) ArticleTitleA bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295 679–682 Occurrence Handle10.1126/science.1067025 Occurrence Handle1:CAS:528:DC%2BD38XptF2rsg%3D%3D Occurrence Handle11809974
H Nagai CR Roy (2001) ArticleTitleThe DotA protein from Legionella pneumophila is secreted by a novel process that requires the Dot/Icm transporter. EMBO J 20 5962–5970 Occurrence Handle10.1093/emboj/20.21.5962 Occurrence Handle1:CAS:528:DC%2BD3MXovVSit7g%3D Occurrence Handle11689436
K O'Donnell HC Kistler BK Tacke HH Casper (2001) ArticleTitleGene phylogenies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc Natl Acad Sci USA 97 7905–7910 Occurrence Handle10.1073/pnas.130193297
M Purcell HA Shuman (1998) ArticleTitleThe Legionella pneumophila icmGCDJBF genes are required for killing of human macrophages. Infect Immun 66 2245–2255 Occurrence Handle1:CAS:528:DyaK1cXivFKqtL4%3D Occurrence Handle9573114
CR Roy RI Isberg (1997) ArticleTitleTopology of Legionella pneumophila DotA: an inner membrane protein required for replication in macrophages. Infect Immun 65 571–578 Occurrence Handle1:CAS:528:DyaK2sXpslKrug%3D%3D Occurrence Handle9009315
CR Roy K Berger RR Isberg (1998) ArticleTitle Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol 28 23–32 Occurrence Handle10.1046/j.1365-2958.1998.00841.x
MM Samrakandi SLG Cirillo DA Ridenour LE Bermudez JD Cirillo (2002) ArticleTitleGenetic and phenotypic differences between Legionella pneumophila strains. J Clin Microbiol 40 1352–1362 Occurrence Handle10.1128/JCM.40.4.1352-1362.2002 Occurrence Handle1:CAS:528:DC%2BD38XjtlCmsrg%3D Occurrence Handle11923356
G Segal M Purcell HA Shuman (1998) ArticleTitleHost cell killing and bacterial conjugation require overlapping sets of genes with a 22-kb region of the Legionella pneumophila genome. Proc Natl Acad Sci USA 95 1669–1674 Occurrence Handle10.1073/pnas.95.4.1669 Occurrence Handle1:CAS:528:DyaK1cXht1Wqtbg%3D Occurrence Handle9465074
G Segal HA Shuman (1999) ArticleTitlePossible origin of the Legionella pneumophila virulence genes and their relation to Coxiella burnetii. Mol Microbiol 33 669–670 Occurrence Handle10.1046/j.1365-2958.1999.01511.x Occurrence Handle1:CAS:528:DyaK1MXlslSju74%3D Occurrence Handle10417657
K Severinov A Mustaev A Kukarin O Muzzin I Bass SA Darst A Goldfarb (1996) ArticleTitleStructural modules of the large subunits of RNA polymerase. J Biol Chem 271 27969–27974 Occurrence Handle10.1074/jbc.271.44.27969 Occurrence Handle1:CAS:528:DyaK28XmvVagurg%3D Occurrence Handle8910400
BJ Stone YA Kwaik (1999) ArticleTitleNatural competence for DNA transformation by Legionella pneumophila and its association with expression of type IV pili. J Bacteriol 181 1395–1402 Occurrence Handle1:CAS:528:DyaK1MXhslWrtrw%3D Occurrence Handle10049368
S Suerbaum J Maynard Smith K Bapumia G Morelli NH Smith E Kunstann et al. (1998) ArticleTitleFree recombination within Helicobacter pylori. Proc Natl Acad Sci USA 95 12619–12624 Occurrence Handle10.1073/pnas.95.21.12619 Occurrence Handle1:CAS:528:DyaK1cXmsl2qtbg%3D Occurrence Handle9770535
MS Swanson BK Hammer (2000) ArticleTitle Legionella pneumophila pathogenesis: a fateful journey from amoebae to macrophages. Annu Rev Microbiol 54 567–613 Occurrence Handle10.1146/annurev.micro.54.1.567 Occurrence Handle1:CAS:528:DC%2BD3cXotFWrsLo%3D Occurrence Handle11018138
DL Swofford (1999) PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer Associates Sunderland, MA
JP Vogel HL Andrews SK Wong RR Isberg (1998) ArticleTitleConjugative transfer by the virulence system of Legionella pneumophila. Science 279 873–876 Occurrence Handle10.1126/science.279.5352.873 Occurrence Handle1:CAS:528:DyaK1cXhtVOisb4%3D Occurrence Handle9452389
A van Belkum M Struelens A de Visser H Verbrugh M Tibaryenc (2001) ArticleTitleRole of genomic typing in taxonomy, evolutionary genetics, and microbial epidemiology. Clin Microbiol Rev 14 547–560 Occurrence Handle10.1128/CMR.14.3.547-560.2001 Occurrence Handle1:CAS:528:DC%2BD3MXlslWlsLs%3D Occurrence Handle11432813
JP Vogel RR Isberg (1999) ArticleTitleCell biology of Legionella pneumophila. Curr Opin Microbiol 2 30–34 Occurrence Handle10.1016/S1369-5274(99)80005-8 Occurrence Handle1:CAS:528:DyaK1MXhsFyiurk%3D Occurrence Handle10047559
WG Weisburg ME Dobson JE Samuel GA Dasch LP Mallavia O Baca et al. (1989) ArticleTitlePhylogenetic diversity of the Rickettsiae. J Bacteriol 171 4202–4206 Occurrence Handle1:CAS:528:DyaL1MXltFaitbY%3D Occurrence Handle2753854
LA Wiater K Dunn FR Maxfield HA Shuman (1998) ArticleTitleEarly events in phagosome establishment are required for intracellular survival of Legionella pneumophila. Infect Immun 66 4450–4460 Occurrence Handle1:CAS:528:DyaK1cXlslCrurg%3D Occurrence Handle9712800
WC Winn (1999) Legionella. PR Murray (Eds) Manual of clinical microbiology, seventh edn. American Society for Microbiology Washington, D.C. 572–582
SD Zink L Pdersen NP Cianciotto YA Kwaik (2002) ArticleTitleThe Dot/Icm type IV secretion system of Legionella pneumophila is essential for the induction of apoptosis in human macrophages. Infect Immun 70 1657–1663 Occurrence Handle10.1128/IAI.70.3.1657-1663.2002 Occurrence Handle1:CAS:528:DC%2BD38XhsFSltLY%3D Occurrence Handle11854262
Acknowledgements
This work was supported by a grant of the Korea Health 21 Research and Development Project, Ministry of Health and Welfare, Republic of Korea (01-PJ10-PG6-01GM03-0002), and in part by the BK21 project for Medicine, Dentistry, and Pharmacy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ko, K., Lee, H., Park, MY. et al. Mosaic Structure of Pathogenicity Islands in Legionella pneumophila . J Mol Evol 57, 63–72 (2003). https://doi.org/10.1007/s00239-002-2452-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-002-2452-8