
Abstract
Gene duplication is commonly regarded as the main evolutionary path toward the gain of a new function. However, even with gene duplication, there is a loss-versus-gain dilemma: most newly born duplicates degrade to pseudogenes, since degenerative mutations are much more frequent than advantageous ones. Thus, something additional seems to be needed to shift the loss versus gain equilibrium toward functional divergence. We suggest that epigenetic silencing of duplicates might play this role in evolution. This study began when we noticed in a previous publication (Lynch M, Conery JS [2000] Science 291:1151–1155) that the frequency of functional young gene duplicates is higher in organisms that have cytosine methylation (H. sapiens, M. musculus, and A. thaliana) than in organisms that do not have methylated genomes (S. cerevisiae, D. melanogaster, and C. elegans). We find that genome data analysis confirms the likelihood of much more efficient functional divergence of gene duplicates in mammals and plants than in yeast, nematode, and fly. We have also extended the classic model of gene duplication, in which newly duplicated genes have exactly the same expression pattern, to the case when they are epigenetically silenced in a tissue- and/or developmental stage-complementary manner. This exposes each of the duplicates to negative selection, thus protecting from “pseudogenization.” Our analysis indicates that this kind of silencing (i) enhances evolution of duplicated genes to new functions, particularly in small populations, (ii) is quite consistent with the subfunctionalization model when degenerative but complementary mutations affect different subfunctions of the gene, and (iii) furthermore, may actually cooperate with the DDC (duplication– degeneration–complementation) process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ever since the publication of a classic book by Ohno (1970), gene duplication has been universally regarded as a main path to evolution of new functions. However, a closer look reveals a difficulty known as the “loss-or-gain” dilemma (Wagner 1998, 2001). For a newly duplicated gene, selection against mutations is relaxed, but a new function gain is only a remote possibility. Accordingly, any mutation that was formerly deleterious in a single-copy gene becomes virtually neutral in its free-evolving duplicate; instead of being eliminated by negative selection, it can be fixed by random drift. Deleterious mutations occur much more frequently than mutations that are advantageous toward the generation of a new function. Thus, one would expect that if selection returns after the duplicate has been protected by redundancy for a long time, it would usually find a nonfunctional pseudogene.
For the loss-or-gain alternative, population genetics theory predicts that, indeed, “pseudogenization” is the most likely evolutionary fate of the vast majority of extra gene copies (Nei and Roychoudhury 1973; Kimura and King 1979; Ohta 1987; Clark 1994; Walsh 1995), especially in species with a relatively small effective population size N e. Consistent with this prediction is an indirect evaluation of pseudogenization rates for young gene duplicates in nine eukaryotic species (Lynch and Conery 2000). The major conclusion of this unprecedented genomewide study is that although gene duplications appear to occur as frequently as single-base substitutions, more than 90% of newly born gene copies degrade to pseudogenes. However, previous estimations made from electrophoretic studies of isozymes at duplicate loci and from the analysis of some mammalian multigene families presumably reflecting ancient polyploidization events in early vertebrate evolution yielded a substantialy higher frequency of adaptive divergence of gene duplicates, about 50% (Nadeau and Sankoff 1997; Wagner 1998). Consistent with the latter, recent direct surveys of “true” (unprocessed) pseudogenes showed that in the 21st and 22nd chromosomes of the human genome these pseudogenes comprise only 4–22% of all identified genes (Harrison et al. 2002). This discrepancy between pseudogenization rates reported by Lynch and Conery (2000) and by Nadeau and Sankoff (1997) as well as the rather low frequency of unprocessed pseudogenes (Harrison et al. 2002) calls for an explanation.
It has recently been realized, however, that the most common immediate fate of duplicated complex genes might be neither nonfunctionalization nor neofunctionalization but rather subfunctionalization, which is defined as a partition of function(s) originally performed by the single ancestral gene between its duplicates. Subfunctionalization can be provided by degenerative but mutually nonoverlapping, complementary mutations in regulatory elements. This DDC (duplication–degeneration–complementation) mutational process (Fig. 1C) either preserves duplicates as such or actually releases a gene from pleiotropic constraints for further evolutionary polishing of the subfunctions (Force et al. 1999; Lynch and Fotce 2000; Lynch et al. 2001).

Models of the evolutionary “loss-or-gain” fate of newly born gene duplicates. It is implied that the extra copy of the gene has not been lost by random drift. Fitnesses of the corresponding mutant alleles are indicated relative to that of the wild-type allele α0: neutral (s = 0), deleterious (s < 0), and advantageous (s > 0), respectively. X denotes a degenerative mutation; *, advantageous mutation. Model A: Both gene duplicates have the same expression pattern (Walsh 1995). Model B: Gene duplicates are silenced (shaded boxes) in the stage (tissue)-differential manner; all other assumptions are the same as in model A. Model C: The small and large boxes denote the regulatory and coding regions, respectively; epigenetically silenced regions are shaded; arrows indicate active states of the duplicates in the corresponding tissues. For simplicity, the case of only two tissue/stage-specific regulatory sequences is shown. Top: The unique original gene with two preexisting tissue/stage-specific regulatory regions (denoted 1 and 2). Middle (DDC): Two possible pairs of complementary degenerative mutations that occurred in nonoverlapping regulatory regions after gene duplication. Bottom (ES + DDC): The same pairs of mutations, but in epigenetically silenced duplicates. In each of two cases of silencing, only one of two pairs of complementary mutations is consistent with the corresponding tissue/stage specificity (a but not b and, symmetrically, d but not c).
We approached the problem of neo- and sub-versus nonfunctionalization from two perspectives, theoretical modeling and genome data analysis. In addition to common models (Fig. 1A), in which just-duplicated genes are indistinguishable in their expression pattern, we considered the model (Fig. 1B) wherein twin genes are active and silent at different developmental stages and/or in different tissues. In this model there is a particular tissue and/or period in development when either of two gene copies is silenced and hence only its active twin is “sensed” by the selection. Epigenetic mechanisms govern such developmental stage- or tissue-specific silencing. Our basic premises are that (i) mutations of any evolutionary significance occur in the germ line, whereas the fitness of corresponding mutant alleles is tested at somatic levels, during ontogenesis, and (ii) newly emerged duplicates may be nonidentical with respect to their epigenetic control. In preservation of gene duplicates, epigenetic silencing may actually “direct” the DDC process by favoring selection of the appropriate pair of degenerative mutations.
As an experimental approach possibly implicating DNA methylation we analyzed two sets of genomewide data on gene duplicates provided by (i) young-vs-old duplicate comparisons (Lynch and Conery 2000) and (ii) catalogs of protein-encoding gene families (Venter et al. 2001). More specifically, we compared the relative expansion of multiprotein families listed by Venter et al. (2001), which are common in five species, Homo sapiens, Arabidopsis thaliana, Drosophila melanogaster, Caenorhabditis elegans, and Sacharomyces cerevisiae.
Both studies reported here indicate that epigenetic silencing of gene duplicates could, indeed, aid evolution toward a new function—but only in populations with a relatively small N e. For the majority of common multiprotein families, their relative expansion appears to have been more efficient in human and plant than in worm and fly genomes—a difference that most likely reflects domain reshuffling and underlying genetic duplications. Consistent with this is a difference we noticed in the data reported by Lynch and Conery (2000): Presumably functional young gene duplicates are definitely more frequent in human, mouse, and the plant Arabidopsis thaliana, all of which have methylayted genomes, than in fly, worm, and yeast genomes, which do not have methylated DNA.
The Evolutionary Advantage of Epigenetic Silencing
Classic Model of Gene Duplicates
Figure 1A shows the classic model of a pair of gene duplicates. For those new duplicates, which are not simply lost by random drift, the model predicts two possible fates. One is mutational degeneration of one of the two copies to a pseudogene; the other is their functional divergence, when advantageous mutations in one of the two genes direct its evolution toward a new function. Functional divergence is often a multistep process. Yet as argued by Walsh (1995), fixation of the first mutation in a gene duplicate is the most critical event. The question is whether a duplicate is first fixed as a null or disadvantageous mutation, thus becoming a pseudogene, or as an advantageous mutation, thus starting the process of functional divergence. For a model of random-mating diploid populations, Walsh (1995) examined the relative probabilities of these two events (denoted in Fig. 1A ψ [null] and f [advantageous]) under a number of simplifying assumptions including the following: (i) lack of linkage, gene conversion and unequal crossing-over; (ii) duplicated loci are initially fixed in a population; (iii) each new mutation is either fixed or lost before the next mutation occurs; and (iv) null mutations at the duplicated gene are neutral and occur at the rate µ per copy per generation, while (v) advantageous mutations are additive with fitnesses 1:1 + s:1 + 2s in the corresponding homo- and heterozygotes (s > 0 is the selection advantage) and occur at the rate µρ, where ρ is the ratio of advantageous to null mutations. It is reasonable to assume that typically ρ ≪ 1. According to standard diffusion theory of population genetics (Crow and Kimura 1970), the expected rates of appearance of successful null and advantageous mutations (destined to be fixed in a population of effective size N e) are ψ = µ and f = µρ [4N e s/(1 − e −4Nes)], respectively (Walsh 1995). Then the probability that the extra gene copy first fixes an advantageous rather than a null mutation is

Fixation of the first advantageous mutation greatly protects a duplicate from the risk of further pseudogenization (Walsh 1995). Indeed, this event reappraises values because it makes the extra gene functional; therefore, any null mutation is no longer neutral, and selection acts against it.
Analysis of Eq. (1) shows that, at given s and ρ values, the potential to evolve toward a new gene is much lower for organisms with a relatively small N e, which is the case of higher organisms with complex, slow development and a long reproductive cycle. This is quite a paradox because even though such processes as alternative splicing can considerably increase a repertoire of functions encoded by a given number of genes, evolution toward more complex development generally implies more functional gene duplicates. The inconsistency becomes more apparent if one considers the following constraint dictated by the general “paradox of rates” (Ratner et al. 1996). Using the theorem about the mean, one can approximate the rate of molecular evolution (Kimura 1971) as

where µ(s) is the probability density for the mutant allele with selective advantage s in a unit of time, p 0 is the original frequency of this allele, u(p 0, s) is the probability of asymptotic fixation of the allele already formed (it is equal to p 0 = 1/(2N e) for neutral mutations), M n and M ad are the summarized probabilities of neutral and adaptive mutations, respectively.
Formula (2) indicates that it is advantageous, not neutral, mutations that distinguish organisms of different N e. According to (2), adaptive evolution of, for example, macroorganisms (with small N e) is expected to be many orders of magnitude slower than that of microorganisms (with incomparably larger N e). The “inequality” becomes of vital significance wherever these “simple” and complex organisms are involved into direct coevolution, e.g., in “parasite–host” systems (Rodin 1991; Ratner et al. 1996). It might be only gene duplications and exon (domain) reshuffling processes that allowed higher organisms to overcome this constraint, in general, and in their immune response against rapidly evolving microorganisms, in particular. The abundance and reshuffling of variable genes of immunoglobulins have been developed by higher organisms as though to compensate for the restricted size of their populations (Rodin 1991; Ratner et al. 1996).
Thus, the “strategic” dilemma for species with not too large N e is as follows: How can natural selection be relaxed in duplicated genes and yet at the same time be on permanent alert to gradually accumulate rare advantageous mutations and eliminate incomparably more frequent deleterious mutations? A possible solution would be some kind of compromise between the single and the duplicated states of genes. In a single gene, the negative selection efficiently eliminates deleterious mutations but the positive selection has no material for a new function. Conversely, gene duplication opens a road toward a new function without losing an old one but the negative selection against deleterious mutations becomes ineffective.
In search of this compromise, we turned to homologous genes in present-day multigene families. These genes (derived from a common precursor by duplication) are usually expressed at different stages of ontogenesis and/or in different tissues and, accordingly, have no problems whatsoever with testing mutations by selection. This stage/tissue specificity in expression might be the very early step in preservation of duplicate genes (Ohno 1970). Indeed, the combined analysis of yeast microarray gene expression data and synonymous substitutions in duplicate genes of different ages indicates that the majority of duplicates seem to have diverged in expression very quickly (Gu et al. 2002a). Not initially specifying possible mechanisms of such stage/tissue-specific silencing events let us formally consider the corresponding model (Fig. 1B).
Stage- and/or Tissue-Complementary Silencing of Twin Genes
Differences between gene duplicates in stage- and tissue-specific expression patterns radically change their fate in favor of functional divergence, including gaining a new function. Comparison of models A and B (Fig. 1) indicates where the change comes from. In model A, alleles α1 and α2 with a deleterious mutation are neutral (s = 0) because each has one functional gene. Consequently, both these alleles can be fixed in a population by random genetic drift. In model B, the same alleles (α1 and α2) become deleterious (s < 0) because for each of them there is a specific developmental stage and/or tissue with the normal gene copy silenced. Accordingly, negative selection will eliminate both of them from a population. The corresponding rate ψ equals 4µN e s/(e 4Ne s − 1), where s < 0 is the selection pressure against a deleterious allele (Crow and Kimura 1970). As for advantageous mutations, alleles α3 and α4 are basically equivalent in models A and B: Compared to the wild-type allele (α0), they both have increased fitness (s > 0) so that positive selection will spread them in a population. The resulting balance in model B is strongly shifted against degeneration into pseudogenes. If, for example, one assumes (for simplicity) equal selection pressure (in its absolute value, |s|) for all four, advantageous and disadvantageous, mutant alleles, then the probability U f for duplicated genes to begin functional divergence instead of becoming a pesudogene is

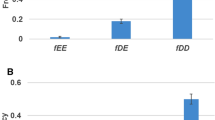
which is much higher than that in model A (compare Eqs. [1] and [3] in Fig. 2)

The effect of silencing on the probability U f that a duplicated gene fixes an advantageous (rather than a degenerative) allele as a function of 4N e s. For simplicity (as in Walsh 1995) the selective disadvantage of deleterious mutations is assumed to be equal in its absolute value, |s|, to the selective advantage of adaptive mutations. A Plots of Eqs. (1) and (3). B The ratio of the U f described by Eq. (3) to that described by the basic Eq. (1). Within the range of relatively small Ne s, this ratio decreases notably with ρ (the two cases shown are for ρ = 0.01 and 0.1).
Mutational and Epigenetic Silencing
In far-diverged homologous genes, stage/tissue-complementary silencing (Fig. 1B) is controlled epigenetically, through the corresponding regulatory sequences. Thus, the whole point of the “loss-or-gain” issue may be concealed not so much in duplicate genes per se as in their regulatory elements. Two possible starts are conceivable for evolution of newly duplicated genes with identical regulatory elements toward stage/tissue-specific expression. First, in accordance with the DDC model (Force et al. 1999; Lynch and Force 2000; Lynch et al. 2001), stage/tissue-specific regulatory regions are silenced by complementary degenerative mutations after duplication (Fig. 1C). As a result, the original function(s) is(are) partitioned between gene duplicates, thus preserving both duplicates with an opportunity for further fine-tuning of the subfunctions in the stage/tissue-specific context. In small populations, if the degenerative mutation rate value per one regulatory element is assumed to be comparable with that per coding region, the DDC process does often lead to the situation shown in Fig. 1B (Force et al. 1999; Lynch and Force 2000; Lynch et al. 2001). Undoubtedly, for newly duplicated genes with multiple subfunctions established earlier, DDC is the most common mechanism of their joint preservation, which is strongly supported by comparative empirical data.
Second, somatic epigenetic silencing (ES) itself is a possibility, through DNA methylation and/or some other mechanisms involving heritable chromatin structure (Jenuwein and Allis 2001). The general ES model is shown in Fig. 1B. It hypothesizes that newly emerged structurally identical duplicates can be nonidentical with respect to their epigenetic control during ontogenesis. There is increasing evidence that stage/tissue-restricted expression of the gene is influenced not only by its direct interplay with numerous transcription factors but also by its chromatin environment within the genome or even within the nucleus (Cockell and Gasser 1999). Apparently, the duplication event itself might essentially change this environment for newly born identical genes. One would expect for position effects of this type to be generally more frequent for translocated rather than tandem duplications. Furthermore, early relocation of tandem gene duplicates to epigenetically different compartments could promote their functional divergence. Some multigene families are intriguing in this regard. For example, α- and β-hemoglobin genes have definitely originated from a common single precursor and were closely linked in an ancestral jawed vertebrate. One would think that tandem organization of genes makes their coordinated regulation easier. However, in birds and mammals α- and β-globin genes lie on separate chromosomes, have contrasting subnuclear environments, chromatin statuses, and methylation levels, and, consequently, exhibit marked tissue-restricted differences in expression (Brown et al. 2001). We suggest that in general such epigenetic differences might favor repositioning of initially tandem gene duplicates.
More specifically, the duplication event might bring one of twin genes farther from (or closer to) stage/tissue-specific regulatory sites such as, for example, methylation-sensitive CpG-containing sequences (Sadhu et al. 1997). Methylation has long been thought to silence genes (Riggs 1975; Holliday and Pugh 1975; Riggs and Porter 1996). For example, a single methylation event at some CpG sites within the CTCF-dependent insulator was shown to be sufficient to inverse the imprinting pattern of the mouse Igf2/H19 locus (Bell and Felsenfeld 2000).
We are unaware of any study aimed at systemic search for stage/tissue-specific methylation-mediated silencing of recently duplicated genes. However, increasing data indicate that, indeed, genetic duplications of different magnitudes (from a separate gene or its parts, to long chromosome segments, to a whole genome) are often accompanied by methylation-mediated inactivation of duplicates (Flavell 1994; Lee and Chen 2001). In fact, for some organisms it is known that duplicates are specifically marked by methylation and efficiently silenced (Rossignol and Faugeron 1994). In plants a transgene often triggers methylating and silencing of the endogenous homologue (Wolffe and Matzke 1999).
Interestingly, ES might to some extent direct DDC. For example, of the two complementary symmetric pairs of degenerative mutations shown in Fig. 1C, only one appears to be selectively valid, namely, the pair of mutations that occurred in regulatory elements compatible with the stage/tissue-specific methylation-silencing pattern (Fig. 1C: variant a or d but not b or c).
A difference between the DDC and the ES models is that DDC implies the preexistence of the gene’s discrete regulatory sequences before the duplication event, whereas this is not necessary for ES. Not much is known with certainty about evolution of “noncoding” regulatory DNA. Yet in paralogous genes it evolves much faster than coding DNA (Harrison, 1998), so it is quite likely that new regulatory sequences can originate at about the time of (or even after) gene duplication, i.e., when an increase in regulatory complexity does not precede but rather accompanies duplication events. One and the same multigene family may show evidence (though indirect) of both degeneration and generation of regulatory regions during evolution (Skaer et al. 2002; Chiu et al. 2002). Virtually nothing is known about the source of new regulatory sequences. Among other possibilities, they, like any piece of DNA, could evolve from duplicates of already existing sequences. If so, we again face the “loss-or-gain” dilemma, readdressed this time from genes as a whole to their regulatory regions. Consequently, to preserve these regions we should, by the DCC mechanism, again suggest the presence of regulatory regions of the higher order (to regulate the regulators), and so on and so forth—which does not really make a lot of sense.
Analysis based on the models shown in Fig. 2 leads to the following conclusions: (i) in very small populations random genetic drift dominates over selection; hence in both systems, with and without epigenetic stage/tissue-complementary silencing of duplicates, pseudogenization is the most likely outcome; (ii) epigenetic silencing does favor evolution of extra genes toward new functions but only in relatively small populations; (iii) in very large populations selection greatly dominates over random drift so that the probability of random fixation of mutations becomes virtually zero; and (iv) the advantage of epigenetic silencing is in its reverse dependence on ρ (the rarer is an advantageous mutation among degenerative ones, the larger is the beneficial effect of epigenetic silencing), thus clearly pointing to the protective role of complementary epigenetic silencing of an extra gene (otherwise it will be inevitably pseudogenized).
In general, one would expect for epigenetic silencing to be more common in organisms with complex development and a small effective population size N e. This correlation does exist for DNA methylation, a known component of epigenetic, relatively stable gene silencing. Mammals and higher plants have highly methylated genomes, whereas organisms with large populations and short generation times, e.g., D. melanogaster, C. elegans, and S. cerevisiae, do not (Tweedie et al. 1997).
At the genome level epigenetic silencing should enhance the functional expansion of multigene families by decreasing degeneration of duplicates, again more efficiently in organisms with a small N e. Consistent with this are the major results of the second part of our study, described below.
Genomewide Data Analysis
Paradox of Young and Old Gene Duplicates
The simple but informative index traditionally used as a cumulative measure of selection pressure on protein-encoding homologous genes is the ratio R/S, where R is the number of observed nonsynonymous base substitutions (resulting in amino acid changes) per replacement site and S is the number of synonymous substitutions per silent site (Li 1997). Since most genes accumulate silent mutations in a time-proportionate manner (Kimura 1983; Li 1997), the S value for each pair of genes is a rough measure of its evolutionary age since their origin by duplication from a common progenitor. R/S = 1 corresponds to neutral evolution with relaxed selection, R/S < 1 reflects the pressure of negative (“purifying”) selection, which preserves an already established “old” function(s), and R/S > 1 indicates a positive selection acting in favor of nonsilent mutations.
Using this criterion, Lynch and Conery (2000) conducted a global analysis of homologous sequences in genome databases for several eukaryotic species. All known or suspected pseudogenes, processed retrogenes, and transposable elements were filtered out of their analyses. To minimize the confusing interference of gene conversions, the authors also excluded all families containing more than five genes (Lynch and Conery 2000). Then, for each pair of presumably functional genes, the R/S ratio was calculated.
The two major conclusions of Lynch and Conery’s study were that there are a very high rate of gene duplication and (at the same time) a very quick degradation of young “extra” genes into pseudogenes. By analyzing their data on the frequencies of young and old duplicates, we found additional contrasts of interest. As Fig. 3A shows, relatively young duplicates (S ≤ 0.1) are notably more frequent in plant, mouse, and human than in fly, worm, and yeast genomes (55–65% vs 7.5–21%, respectively). Remarkably, the reverse is the case for old duplicates: their frequency is higher in invertebrates than in plants and mammals (Fig. 3A). If, as Fig. 3A suggests, genes duplicate much less frequently in yeast, worm, and fly, where did their more frequent old duplicates come from? Similarly, if genes duplicate much more frequently in plant, mouse, and human, but old duplicates are rare, why do these young extra genes disappear? The simplest explanation would be that compared to yeast and invertebrates, genes in plants and mammals do duplicate more frequently but then degenerate even more rapidly into pseudogenes. To test the hypothesis, one needs to perform a parallel similar comparison for pseudogenes. Unfortunately, this is difficult to do because identification of unprocessed pseudogenes is imprecise (Mounsey et al. 2002). As far as reliably identifiable pseudogenes are concerned, they were disregarded by Lynch and Conery (2000).

The paradox of young and old duplicates. A Frequency distributions of pairs of homologous genes as a function of evolutionary age as measured by S, the number of silent substitutions per silent site (adopted from Lynch and Conery 2000). The distributions for six eukaryotic species are shown: Homo sapiens (H), Mus musculus (M), the plant Arabidopsis thaliana (P), the fly Drosophila melanogaster (F), the worm Caenorhabditis elegans (W), and the yeast Saccharomyces cerevisiae (Y). Relatively young duplicates are in the range of S ≤ 0.1; definitely old duplicates (with S varying in the 1.5–3.0 range) are grouped together. B Frequency distributions of protein family size for the same species as in A, except for M. musculus; theirpairwise representations are shown in Fig. 6 (see text for details).
There is, however, another reasonable explanation for the paradox, which we favor and which, in its extreme form, is opposite to the previous one. We propose that genes from the two groups might differ not only (and not so much) in their duplication rate but in the rate of their degeneration, i.e., plant, mouse, and human gene duplicates might be better protected from pseudogenization than their yeast, nematode, and fly homologues. This actually means that during any amount of time the probability of a young duplicate becoming new functional gene and thus increasing the size of the family to which it belongs is higher in plants and mammals than in yeast and invertebrates. If so, a considerable fraction of young duplicates in plant and mammalian genomes (Fig. 3A) might not have degenerated at all but, rather, “moved up” to a new function. Naturally, such a transition is not seen in Fig. 3A, wherein only families with a small number (five or fewer) of genes were analyzed (Lynch and Conery 2000). Our study of the relative expansion of protein-encoding genes (see below) fills the gap and supports the move-up hypothesis.
Expansion of Protein Families in Different Eukaryotic Species
The current interest in interspecies comparative studies of proteomes is generally focused on the protein-encoding multigene families that distinguish one species from the others, with a natural bias to human-specific genes (Venter et al. 2001). Contrary to that strategy, we elected to focus on the species-common families. As primary data, we used Table 18 of Venter et al. (2001), which represents the Pfam domain-based catalog of proteins with known function(s) in five sequenced genomes, yeast (S. cerevisiae), plant (A. thaliana), invertebrates (the worm C. elegans and the fly D. melanogaster), and vertebrates (H. sapiens). Moreover, we have selected only the families that contained at least two genes in each of the five species to ensure exclusion of the cases where ancestral genes, for species-specific reasons, either lost the ability to duplicate or never had it. The resulting list contains 57 common families. Some of the genes certainly code for mosaic proteins of the complex domain architecture containing more than one domain type. Genes encoding these mosaic proteins most likely originate from exon reshuffling. It is clear that the exon reshuffling-based origin of new genes (without losing the old ones) necessarily suggests duplication of the corresponding constituents. Each of these particular families requires a separate interspecies comparative analysis. In general, however, according to calculations by Li and co-workers (2001), the mosaic part of proteomes does not vary as much between the species as one might expect: 28, 27, 21, and 19% in the H. sapiens, D. melanogaster, C. elegans, and S. cerevisiae genomes, respectively. This means that a significant difference in the expansion of homologous proteins between species (if detected) should reflect their difference not only in primary duplication rate but also in functional divergence of emerged gene copies.
Interspecies comparisons of these 57 gene families revealed some general patterns, shown in Figs. 4 and 5. The yeast genome was significantly inferior in the number of homologous genes—only in 10 of 228 cases does yeast outnumber other species (not shown). On visual inspection of Fig. 4, it is apparent, for example, that more family members are found in human than Drosophila (Fig. 4A). To confirm and extend visual inspection, the one-way correlated-samples ANOVA test (with subsequent Tukey HSD test) was carried out for comparison of the four species. Three tests were performed—for all 57 gene families, for 45 families containing no more than 100 genes for either species, and for 33 families containing no more than 50 genes for either species. The rationale behind this partitioning is that (i) many large families actually represent very complex superfamilies and (ii) species-specific selection was certainly involved in their expansion (Venter et al. 2001); therefore data sets restricted to the smaller gene families should prove more revealing for our purposes. All three ANOVA tests were (nonsurprisingly) highly significant (at the <0.01 level), thus rejecting the null hypothesis (that all the population means are equal). Once the null hypothesis was rejected, it was of interest to determine where exactly the differences lie by performing pairwise comparison tests. For that purpose we used a comparatively conservative Tukey’s HSD (honestly significantly different) test to keep the EER (experimentwise error rate) well under control, even if at the expense of trading off some power. Therefore, the following results should be interpreted as conservative. Turkey’s HSD test for the smallest data set (no more than 50 genes in each gene family for each species) was not significant for human-vs-plant and fly-vs-worm comparisons and highly significant (at the <0.01 level) for the remaining four comparisons. Tukey HSD test for the intermediate data set was not significant for human-vs-plant and fly-vs-worm comparisons, highly significant (at the <0.01 level) for human-vs-fly and human-vs-worm comparisons and significant (at the <0.05 level) for plant-vs-fly and plant-vs-worm comparisons. The test for the complete set was significant for human-vs-fly (at the <0.01 level) and human-vs-worm (at the <0.05 level) comparisons and not significant for all remaining comparisons.

The relative expansion of 57 common protein families in the human, fly, worm, and plant genomes. The primary data are from Venter et al. (2001). Shown are six pairwise interspecies comparisons. Almost each point corresponds to a single family; cases where different families have exactly the same size in both species are very rare. The comparisons have not been normalized for species-specific proteome sizes. The cases of two species having equal numbers of genes map along the diagonal. Sectors with ≤5 genes are outlined.
These results clearly indicate that the four species are divided into two distinct groups (Fig. 4) that are in fact the same as in Fig. 3A. The human genome is certainly superior to the fly and worm genomes with regard to genes per family (Fig. 4A and B). Strikingly, A. thaliana demonstrates the same tendency in comparisons with D. melanogaster and C. elegans (Fig. 4C and D). In particular, almost all families that in D. melanogaster and C. elegans contain no more than five genes are mapped above the diagonal, i.e., their orthologous counterparts are more expanded in H. sapiens and A. thaliana (Fig. 4A–D), thus explaining the paradox of young and old gene duplicates (Fig. 3A). At the same time, neither the H. sapiens-vs-A. thaliana (Fig. 4E) nor the D. melanogaster-vs-C. elegans (Fig. 4F) comparison showed evidence of one species outrunning the other. The average range of family member expansion is, however, obviously wider in the human-vs-plant than in the fly-vs-worm comparison.
Consistent with this are interspecies pairwise comparisons of the general family size distribution (Fig. 5). These distributions also divide the species into the same two groups. The predominant fraction of gene families in fly and worm contains a small number (2–4) of genes, whereas the gene families from human and plant genomes are distributed more evenly within the range of 4–20 genes. Again, neither the human-vs-plant (Fig. 5E), nor the fly-vs-worm (Fig. 5F) comparison revealed any notable difference in the distribution of family size.

Frequency distributions of protein family size. The primary data are from Venter et al. (2001). All large families (with more than 80 proteins) are grouped together. In each comparison, thenumber of common families is indicated.
The majority of the species-common 57 families represent genes with an already established function(s) (Venter et al. 2001). Although species-specific expansion is more frequent among large families, it is also noticeable in the small and medium size range (Fig. 4). Undoubtedly, a major force that drives the expansion of these individual genes is species-specific natural selection (Venter et al. 2001). Some apparent examples are immunoglobulin domains in H. sapiens, S/T and dual-specificity protein kinase in A. thaliana, and the F-box domain in C. elegans (Venter et al. 2001). However, as Fig. 4 clearly indicates, the general trend is that most of the gene families have gained more new functional members in H. sapiens and A. thaliana than in D. melanogaster and C. elegans. The difference is especially surprising if one takes into account that (i) the effective size N e of the human population is certainly smaller than that of fly and nematode and, on the contrary, (ii) the generation time is larger in human and mustard weed than in fly and worm.
We hypothesize that the differences between these two groups (Figs. 3,4,5) reflect not only species-specific selection (Venter et al. 2001), primary duplication rates (Lynch and Conery 2000; Li et al. 2001; Gu et al. 2002b), and/or not-so-ancient vertebrate- and plant-specific whole-genome doublings (Ohno 1970; see also Hughes [1999] and Wolfe [2001] for ongoing debate) but rather some underlying mechanism or mechanisms that make the positive selection of new genes more effective in A. thaliana. M. musculus, and H. sapiens than in S. cerevisiae, C. elegans, and D. melanogaster. Quite convincing in this regard is one particular difference between the two groups that we noticed in Fig. 1 of Lynch and Conery’s (2000) article: Consider very recent duplications with S ≤ 0.01 that are therefore apparently not associated with paleopolyploidization events (Wolfe 2001). These “young” duplicates appear to be incomparably more frequent in H. sapiens, M. musculus, and A. thaliana than in D. melanogaster, C. elegans, and S. cerevisiae (see Fig. 1 of Lynch and Conery [2000] for details).
For relatively young duplicates (0 < S < 0.25), Lynch and Conery (2000, 2001) estimated the rate of their loss by using the survivorship function, N S = N 0 e −dS, where N S is the number of duplicates observed at divergence level S. However, the relative expansion of gene families (Fig. 4) indicates that this formula may actually reflect not only the exit of duplicates into pseudogenes as stated by Lynch and Conery (2000) but also their adaptive functional diversification. At the level of general frequency distribution of family size, this process appears to be a transition of duplicates from one family into the next one of larger size (Fig. 5). This explains why the family-size frequency pattern of common families (Fig. 3B) looks like an almost-ideal mirror image of the age-dependent frequency pattern of gene duplicates (Fig. 3A).
Similar interspecies comparisons of domain expansion (not shown) revealed exactly the same two distinct groups of species, consistent with a small variation of the portion of complex genes coding for mosaic proteins in different species (Li et al. 2001). This also supports our original assumption that the interspecies difference in the expansion of homologous proteins would in large part reflect gene duplications.
Taken together, Figs. 3,4,5 indicate that gene duplicates in such distant species as the plant A. thaliana and the mammals M. musculus and H. sapiens have something in common: Compared to S. cerevisiae, C. elegans, and D. melanogaster, they are likely better protected from degeneration to pseudogenes. As mentioned previously, we think it noteworthy that the genomes of H. sapiens, M. musculus, and A. thaliana are methylated, whereas the genomes of D. melanogaster, C. elegans, and S. cerevisiae are not—the difference expected if ES, in addition to DDC, does protect duplicate genes from pseudogenization.
Conclusion
In essence, our model of stage/tissue-complementary ES of gene duplicates combines the evolutionary advantages of two states of a gene—when it is single and when it is duplicated. In the single state, selection purifies from frequent degenerative mutations, while in the duplicated state selection of rare advantageous mutations drives evolution toward a new gene without losing an old one. In fact, the ES model adds another level of complexity to the DDC model. In preservation of gene duplicates with already adjusted regulatory regions, ES plays a subsidiary role compared to the DDC mechanism; however, we feel that this role may become of the first order when these regulatory regions are just in the making. Whatever the actual mechanism(s) of silencing is, mutational and/or epigenetic, it strongly protects genomes from pseudogenization, and this is likely to be especially true for species of a relatively small effective population size and long generation cycle.
The possible role of ES in evolution by gene duplication can be verified and further investigated along the different lines of research. First, a complete and explicit analytical description of epigenetically silent duplicates combined with Monte Carlo simulations is required to estimate the fate of gene duplicates for different duplication events (from local tandem duplications to polyploidization), classes of mutations, linkage degrees, mechanisms of silencing, and selection. Evolutionarily reversible inactivation when a duplicate is “sleeping” for many generations and occasionally “awakens” is of particular interest since such transgenerational ES allows natural selection to eliminate degenerative mutations in the expressed gene while keeping its transiently silenced, but reactivatable, copy able to gain advantageous mutations. This analysis will be published separately. Second, the comparative genomewide analysis should be updated with the data from forthcoming complete genome sequencing of new species and focused on genomes with a different degree of methylation. Third, this analysis should not be limited to multigenic families of a small size (fewer than five members). Finally, in addition to functional members of multigenic families, the relative expansion of reliably identified unprocessed pseudogenes (i.e., junk rather than selfish DNA) should be compared in different species. These studies are under way.
References
AC Bell G Felsenfeld (2000) ArticleTitleMethylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405 482–485 Occurrence Handle10.1038/35013100 Occurrence Handle1:CAS:528:DC%2BD3cXjslykuro%3D Occurrence Handle10839546
KE Brown S Amoils JM Horn VJ Buckle DR Higgs M Merkenschlager AG Fisher (2001) ArticleTitleExpression of α-and β-globin genes occurs within different nuclear domains in haemopoetic cells. Nature Cell Biol 3 602–606 Occurrence Handle1:CAS:528:DC%2BD3MXksFelurc%3D
C-h Chiu C Amemiya K Dewar C-B Kim FH Ruddle GP Wagner (2002) ArticleTitleMolecular evolution of the HoxA cluster in the three major gnathostome lineages. Proc Natl Acad Sci USA 99 5492–5497 Occurrence Handle10.1073/pnas.052709899 Occurrence Handle1:CAS:528:DC%2BD38XjtFKltrw%3D
AG Clark (1994) ArticleTitleInvasion and maintenance of a gene duplication. Proc Natl Acad Sci USA 91 2950–2954 Occurrence Handle1:CAS:528:DyaK2cXktVantrs%3D Occurrence Handle8159686
M Cockell SM Gasser (1999) ArticleTitleNuclear compartments and gene regulation. Curr Opin Genet Dev 9 199–205 Occurrence Handle1:CAS:528:DyaK1MXisFyqtLk%3D Occurrence Handle10322139
J Crow M Kimura (1970) Introduction to population genetics theory. Harper and Row New York
RB Flavell (1994) ArticleTitleInactivation of gene expression in plants as a consequence of specific sequence duplication. Proc Natl Acad Sci USA 91 3490–3496 Occurrence Handle1:CAS:528:DyaK2cXktVajsLo%3D Occurrence Handle8170935
A Force M Lynch B Pickett A Amores Y-l Yan J Postlethwait (1999) ArticleTitlePreservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531–1545 Occurrence Handle1:CAS:528:DyaK1MXisV2rs7o%3D Occurrence Handle10101175
Z Gu D Nicolae H Lu W-H Li (2002a) ArticleTitleRapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet 18 609–613 Occurrence Handle1:CAS:528:DC%2BD38XoslCgur0%3D
Z Gu A Cavalcanti F-C Chen P Bouman W-H Li (2002b) ArticleTitleExtent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol Biol Evol 19 256–262 Occurrence Handle1:CAS:528:DC%2BD38XitFSnsL4%3D
R Hardison (1998) ArticleTitleHemoglobin from bacteria to man: Evolution of different patterns of gene expression. J Exp Biol 201 1099–1117 Occurrence Handle1:CAS:528:DyaK1cXjvVOhsbg%3D Occurrence Handle9510523
PM Harrison A Kumar N Lang M Snyder M Gerstein (2002) ArticleTitleA question of size: The eukaryotic proteome and the problems in defining it. Nucleic Acids Res 30 1083–1090 Occurrence Handle1:CAS:528:DC%2BD38XitVOisLc%3D Occurrence Handle11861898
R Holliday JE Pugh (1975) ArticleTitleDNA modification mechanisms and gene activity during development. Science 187 226–232 Occurrence Handle1:CAS:528:DyaE2MXht1Sitrw%3D Occurrence Handle1111098
AL Hughes (1999) ArticleTitlePhylogenies of developmentally important proteins do not support the hypothesis of two rounds of genome duplication early in vertebrate history. J Mol Evol 48 565–576 Occurrence Handle1:CAS:528:DyaK1MXisFGis7o%3D Occurrence Handle10198122
T Jenuwein CD Allis (2001) ArticleTitleTranslating the histone code. Science 293 1074–1080 Occurrence Handle1:CAS:528:DC%2BD3MXmtVWltro%3D Occurrence Handle11498575
M Kimura (1971) ArticleTitleTheoretical foundation of population genetics at the molecular level. Theor Popul Biol 2 174–208 Occurrence Handle1:STN:280:CSyB2cbntVA%3D Occurrence Handle5162686
M Kimura (1983) The neutral theory of molecular evolution. Cambridge University Press Cambridge
M Kimura JL King (1979) ArticleTitleFixation of a deleterious allele at one of two “duplicate” loci by mutation pressure and random drift. Proc Natl Acad Sci USA 76 2858–2861 Occurrence Handle1:STN:280:CSaB38zovV0%3D Occurrence Handle288072
H-S Lee Z Chen (2001) ArticleTitleProtein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc Natl Acad Sci USA 98 6753–6758 Occurrence Handle1:CAS:528:DC%2BD3MXksVOku7w%3D Occurrence Handle11371624
W-H Li (1997) Molecular evolution. Sinauer Associates Sunderland, MA
W-H Li Z Gu H Wang A Nekrutenko (2001) ArticleTitleEvolutionary analysis of the human genome. Nature 409 847–849 Occurrence Handle1:CAS:528:DC%2BD3MXhsFCjtL4%3D Occurrence Handle11237007
M Lynch JS Conery (2000) ArticleTitleThe evolutionary fate and consequences of duplicate genes. Science 290 1151–1155 Occurrence Handle10.1126/science.290.5494.1151 Occurrence Handle1:CAS:528:DC%2BD3cXotVChsb8%3D Occurrence Handle11073452
M Lynch A Force (2000) ArticleTitleThe probability of duplicate gene preservation by subfunctionalization. Genetics 154 459–473 Occurrence Handle1:CAS:528:DC%2BD3cXms1KhsA%3D%3D Occurrence Handle10629003
M Lynch M O’Hely B Walsh A Force (2001) ArticleTitleThe probability of preservation of a newly arisen gene duplicate. Genetics 159 1789–1804
A Mounsey P Bauer IA Hope (2002) ArticleTitleEvidence suggesting that a fifth of annotated Caenorhabditis elegans genes may be pseudogenes. Genome Res 12 770–775 Occurrence Handle1:CAS:528:DC%2BD38XjvFSgtL0%3D Occurrence Handle11997343
JH Nadeau D Sankoff (1997) ArticleTitleComparative rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147 1259–1266 Occurrence Handle1:STN:280:DyaK1c%2FktlCitA%3D%3D Occurrence Handle9383068
M Nei AK Roychoudhury (1973) ArticleTitleProbability of fixation of nonfunctional genes at duplicate loci. Am Nat 107 362–372
S Ohno (1970) Evolution by gene duplication. Springer Berlin
T Ohta (1987) ArticleTitleSimulating evolution by gene duplication. Genetics 115 207–213 Occurrence Handle1:STN:280:BiiC28zksVM%3D Occurrence Handle3557113
VA Ratner AA Zharkikh NA Kolchanov SN Rodin VV Solovyov AS Antonov (1996) Molecular evolution. Springer Berlin
AD Riggs (1975) ArticleTitleX-inactivation, differentiation and DNA methylation. Cytogenet Cell Genet 14 9–25
AD Riggs TN Porter (1996) Overview of epigenetic mechanisms. EA Russo RA Martienssen AD Riggs (Eds) Epigenetic mechanisms of gene regulation. Cold Spring Harbor Laboratory Press New York 29–45
SN Rodin (1991) Idea of coevolution. Nauka Novosibirsk (in Russian)
JL Rossignol G Faugeron (1994) ArticleTitleGene inactivation triggered by recognition between DNA repeats. Experientia 50 307–317 Occurrence Handle1:CAS:528:DyaK2cXjtFKktb0%3D Occurrence Handle8143804
A Sadhu ML Shen M Hackbarth E Hume TW McKeithan (1997) ArticleTitleCpG-rich sequences close to the site of duplication within the human IGH constant region. Immunogenetics 45 365–370 Occurrence Handle1:CAS:528:DyaK2sXjt12ltrs%3D Occurrence Handle9089093
N Skaer D Pistillo J-M Gibert P Lio C Wulbeck P Simpson () ArticleTitleGene duplication at the achaete-scute complex and morphological complexity of the peripheral nervous system in Diptera. Trends Genet 18 399–405
S Tweedie J Charlton V Clark A Bird (1997) ArticleTitleMethylation of genomes and genes at the invertebrate-vertebrate boundary. Mol Cell Biol 17 1469–1475 Occurrence Handle1:CAS:528:DyaK2sXhtlGmsLg%3D Occurrence Handle9032274
JC Venter et al. (2001) ArticleTitleThe sequence of the human genome. Science 21 1304–1351
A Wagner (1998) ArticleTitleThe fate of duplicated genes: loss or new function? BioEssays 20 785–788 Occurrence Handle10.1002/(SICI)1521-1878(199810)20:10<785::AID-BIES2>3.0.CO;2-M Occurrence Handle1:STN:280:DyaK1M3gs1emug%3D%3D Occurrence Handle10200118
A Wagner (2001) ArticleTitleBirth and death of duplicated genes in completely sequenced eukaryotes. Trends Genet 17 237–239 Occurrence Handle1:CAS:528:DC%2BD3MXjtFCltbo%3D Occurrence Handle11335019
JB Walsh (1995) ArticleTitleHow often do duplicated genes evolve new functions? Genetics 139 421–428 Occurrence Handle1:STN:280:ByqB38%2FmvVQ%3D Occurrence Handle7705642
KH Wolfe (2001) ArticleTitleYesterday’s polyploids and the mystery of diplodization. Nature Rev Genet 2 333–341 Occurrence Handle1:CAS:528:DC%2BD3MXjtlGjs7g%3D
AP Wolffe MA Matzke (1999) ArticleTitleEpigenetics: Regulation through repression. Science 286 481–486
Acknowledgements
We are very thankful to Andrew Rodin for valuable suggestions and help with the statistical treatment of data. This work was supported by NIH Grant GM 50575.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rodin, S.N., Riggs, A.D. Epigenetic Silencing May Aid Evolution by Gene Duplication . J Mol Evol 56, 718–729 (2003). https://doi.org/10.1007/s00239-002-2446-6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-002-2446-6