
Abstract
Objective
This study was designed to characterize the population pharmacokinetics of pyrazinamide in South African pulmonary tuberculosis patients, with special reference to interindividual and interoccasional variability (IIV and IOV, respectively).
Methods
Concentration-time measurements obtained from 227 patients receiving oral doses of pyrazinamide were pooled to create a dataset containing 3,092 data points spanning multiple dosing occasions. The software program NONMEM was used to analyze the data.
Results
A one-compartment model with first-order absorption, including a zero-order component describing release from formulation, and first-order elimination best described the data. The absorption rate constant was estimated to be bimodally distributed between two distinct subgroups, fast and slow, in approximately even proportion. Absorption rate was threefold greater in fast absorbers (3.56 h−1) in comparison to slow absorbers (1.25 h−1). Typical values of oral clearance and apparent volume of distribution were estimated as 3.42 L h−1 and 29.2 l, respectively. IOV was supported in oral clearance (0.0238, variance) and absorption rate (0.623, variance). The duration of zero-order absorption was estimated as 0.290 h, and was quite variable between patients (0.957, variance).
Conclusion
The absorption of pyrazinamide in the studied population was highly variable and two separate subpopulations were identified. IOV accounted for a proportion of the variability in clearance and the absorption rate constant.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pyrazinamide (PZA) is a synthetic analogue of nicotinamide (vitamin B3), and is bactericidal against Mycobacterium tuberculosis [22], acting most effectively against slowly or intermittently metabolizing semi-dormant organisms within macrophages [5]. It is an essential component of tuberculosis combination chemotherapy and is used during the initial 2 months of treatment for its sterilizing activity [11]. The activity of PZA relies on an acid pH for conversion to its primary metabolite, pyrazinoic acid (POA) [10, 13, 17, 23]. PZA is converted to POA by bacterial pyrazinamidase, independently of the host’s metabolism [7]. Its minimum inhibitory concentration (MIC) at pH 5.95 has been shown to be as much as 8-fold lower than its MIC at pH 5.5 [13].
South African tuberculosis treatment guidelines at the time the study was carried out [18] recommended a PZA-containing regimen administered daily, 5 times per week (Monday to Friday) during the intensive phase of treatment (the first 8–12 weeks). In addition to PZA, standard combination tuberculosis therapy included isoniazid (INH), rifampicin (RIF), ethambutol (EMB) and, in cases of retreatment or drug resistance, streptomycin (SM). The population pharmacokinetics of PZA have previously been described in North American healthy volunteers [12] and patients [24]. Pulmonary tuberculosis patients in the developed world are, however, demographically dissimilar to analogous populations in other regions. This study was designed to characterize the drug’s pharmacokinetics in a South African population, with a specific view towards investigating interindividual and interoccasional variability (IIV and IOV, respectively). Such differences in exposure may reflect differences in effect, which may in turn have clinical importance.
Methods
Patients
Data from three unpublished clinical studies in pulmonary tuberculosis patients were pooled to generate the dataset used in the analysis (Table 1). Hospitalized patients were recruited from two treatment centres: the DP Marais SANTA (South African National Tuberculosis Association) Centre, near Cape Town, and Brewelskloof Hospital, in the more rural Breede River Valley in South Africa’s Western Cape province. The study participants were males and non-pregnant females over the age of 18 years. Treatment was administered in accordance with the World Health Organization (WHO)’s Directly Observed Treatment (Short Course) (DOTS) tuberculosis control strategy [8]. PZA was administered in combination with at least RIF and INH, and with EMB and SM as appropriate with respect to the WHO’s tuberculosis treatment recommendations [21] and national guidelines [4]. All subjects were fasted from 2200 hours on the evenings prior to blood sampling, and provided full written informed consent for participation in the studies. Ethical approval for all the studies was granted by the research ethics committees of the University of Cape Town, South Africa and the participating study centers. A total of 3,092 concentration-time observations gathered from 227 subjects were available for modelling. In total, 27 discrete dosing occasions were present in the pooled dataset, covering a cumulative 6-week period measured from the start of blood sampling for PZA pharmacokinetics.
The first subset, which we shall refer to as DPM1, was composed of 56 pulmonary tuberculosis patients. Thirty patients received 1,200 mg/day of PZA orally (Rifafour e-200, Aventis Pharma, Midrand, South Africa), 25 patients received 1,500 mg/day of PZA orally (Rifafour e-200 or Pyrazide 500, Hoechst Marion Roussel, Midrand, South Africa), and 1 patient received 2,000 mg/day of PZA orally (Pyrazide 500) Monday to Friday for a minimum of 2 weeks prior to pharmacokinetic assessment. Sets of three blood samples for the determination of PZA pharmacokinetics were taken twice weekly, on Tuesdays and Fridays, at random times between 0 and 12 h post-dose, for 2 weeks. No changes were made to patients’ prescribed antimycobacterial regimens, which included PZA at the described dosages.
The second subset (DPM2) was composed of 28 pulmonary tuberculosis patients on regular tuberculosis treatment, including PZA, for at least 2 weeks prior to the study. Subjects in this group were excluded in cases of previous hepatic, renal, or gastrointestinal disorder. Subjects with a recent history of concomitant illness, blood donation, or substance abuse were excluded. All patients weighed 50 kg or more, and each received 1,500 mg/day of PZA (Rifcin, Novartis, Isando, South Africa, or Rozide 500, Rolab, Kempton Park, South Africa), in addition to their regular doses of RIF and INH, orally from Monday to Friday. Blood samples for pharmacokinetics were taken pre-dose, and subsequently at 0.25, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, 12.0 and 24.0 h post-dose on Tuesdays on up to 5 occasions. Patients in DPM2 received their other prescribed medications as usual except for the days of pharmacokinetic blood sampling, upon which only PZA, RIF and INH were administered.
The third and final subset of patients (BKH) contained 144 pulmonary tuberculosis patients, admitted at the end of the 2-month intensive phase of treatment which included PZA. Patients were dosed according to the attending physicians’ best clinical judgment, in line with treatment policy at the site. Six patients received 1,000 mg/day of PZA orally (Pyrazide 500), 102 patients received 1,500 mg/day of PZA orally (Isopas 500, Lennon Medicines, Gallo Manor, South Africa; Pyrazide 500, Rozide 500, and Rolab Pyrazinamide, Rolab), and 36 patients received 2,000 mg/day of PZA orally (Isopas 500, Pyrazide 500, Rozide 500 and Rolab Pyrazinamide) daily. In contrast to the 5-day dosing cycles used in the DPM1 and DPM2 studies, BKH patients were administered PZA on 7 days per week as a consequence of differences in treatment policy at the Brewelskloof site. Blood samples for the determination of PZA pharmacokinetics were taken on a single study day (Thursday) pre-dose and subsequently at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0 and 8.0 h post-dose. In addition to PZA, patients received their other prescribed medications as usual.
Covariate information was recorded for each patient by interview and by examination of their clinical case records, and included age, weight, body mass index (BMI), and gender (Table 1). Patient use of alcohol, tobacco, and drugs of abuse were determined by questionnaire. Details of concomitant medication were recorded (none had previously been reported to interact with PZA), and full medical histories were taken from every subject. Compliance on study days was monitored by direct observation.
Specimen collection and storage
Blood samples for determination of PZA pharmacokinetics were collected into 4-ml lithium heparin vacuum tubes (Vacuette, Greiner Bio-One International, Kremsmuenster, Austria) through an intravenous cannula (Introcan 1.1×32 mm, B. Braun, Melsungen, Germany) inserted into an arm vein. The samples were stored temporarily in darkness on ice, before undergoing centrifugation at 3,500 rpm for 10 min. The plasma was subsequently harvested into labelled 1.5-ml microcentrifuge tubes (Greiner Bio-One International, Kremsmuenster, Austria) and stored at −80°C until analysis.
Serum chemistry (urea, creatinine, total bilirubin, alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase) and complete blood cell count (haemoglobin, red cell count, white cell count, mean corpuscular volume and haematocrit) were determined in all participants, for investigation as potential covariate predictors of pharmacokinetic parameters. In addition, patients were requested to submit to testing for the presence of human immunodeficiency virus (HIV) antigens or antibodies, on a voluntary basis, in order to have HIV status available for investigation as a covariate predictor. An automated enzyme-linked immunosorbent assay (ELISA)-based method (AxSYM HIV Ag/Ab Combo, Abbott Diagnostics, Germany) was used for initial analysis of samples. Counseling was provided pre- and post-test, regardless of the result. Manual confirmatory testing was carried out in subjects whose initial screening result was positive (Enzygnost Anti-HIV 1/2 Plus, Dade Behring, Liederbach, Germany).
Drug quantification
Plasma concentrations of PZA were determined by high-performance liquid chromatography (HPLC) using UV detection [16]. The HPLC system consisted of a Spherisorb C8 analytical column of dimensions 25×4.6 mm and particle diameter 5 μm (PSS831815, Waters, Milford, Mass., USA) in conjunction with a reverse-phase guard column (2.5×0.46 cm, packed with Pelliguard LC-8, Upchurch Scientific, Oak Harbor, Wash., USA). The mobile phase consisted of acetonitrile (BDH, Poole, United Kingdom) and 0.06% trifluoroacetic acid (Riedel-de Haën, Seelze, Germany) in the ratio 5:95. The UV detection wavelength was 270 nm and the flow rate was 1.5 ml min−1.
Frozen plasma samples were allowed to thaw in a water bath at ambient temperature. The C18 extraction column (Bond Elut 3.0 cm; Varian, Palo Alto, Calif., USA) was primed with 1 ml 0.5 mM potassium phosphate buffer (pH 4.5) and 0.5 ml of plasma was applied to the column. The sample was drawn onto the column and allowed to stand for 10 min.. The column was washed with 1 ml of the phosphate buffer. Finally, PZA was eluted into an analytical vial using 0.5 ml acetonitrile and 0.5 ml methanol. All solvents were of HPLC grade. Twenty μl of extracted sample was injected directly onto the column at ambient temperature and the retention time was approximately 5.7 min.
The standard curve was linear and provided a detection range of 0.4–70 mg l−1. Quality control samples of 1.0 mg l−1, 30 mg l−1 and 70 mg l−1 were interspersed between the samples. Interday and intraday percentage coefficients of variation (CV) were well below 10% for the low, medium and high quality control samples. Mean (±SD) recovery was assessed by measuring the PZA concentration obtained from spiked plasma samples corresponding to points on the standard curve, and was determined to be 75.8%±5.07%.
Pharmacokinetic data analysis
Model-building was conducted using NONMEM (version V, double precision, level 1.1) [2], implemented on a computer workstation running Mandrake Linux 9.1 (Mandrakesoft, Paris, France) with GNU Fortran (g77) 3.3.2, a component of GCC 3.3.2 (Free Software Foundation, Boston, USA). Model-building steps and associated analysis data were managed using the software utilities Census [20] and Xpose 3.102 [6]. Initially, NONMEM’s first-order conditional estimation (FOCE) with ɛ-η interaction method was used for model development. However, limitations in computer time and difficulties in obtaining covariance information using this method made it necessary to replace it with the first-order estimation (FO) method for the estimation of typical population pharmacokinetic parameters, random variability (IIV and IOV) in these parameters, and residual variability between observed and predicted plasma concentrations.
The data were fitted using one- and two-compartment models with first-order absorption and elimination. Several approaches to modelling absorption were tested, including the use of absorption lag time, combined first- and zero-order absorption models, and the use of a zero-order input into the absorption compartment to mimic the release of drug from solid-phase formulation. Elimination was assumed to take place from the central plasma compartment in all models tested. Bimodal distribution of the rate of absorption, observed in individual plots of the concentration-time data, was investigated using a mixture model. The influence of covariances between model variance parameters was tested through the estimation of off-diagonal elements in the variance-covariance matrix.
IIV, where employed, was expressed in the same manner as shown for oral clearance in Eq. (1):
(CL/F) i is oral clearance for the ith individual, TV(CL/F) is the typical value of oral clearance in the population. \({\left( {\eta _{i} } \right)}_{{CL/F}}\) is the IIV, normally distributed with mean 0 and variance \({\left( {\omega ^{2} } \right)}_{{CL/F}}\), which describes the difference between individual i’s oral clearance and the predicted typical value. IOV, \({\left( {\kappa _{i} } \right)}_{{CL/F}}\), was normally-distributed with mean 0 and variance \({\left( {\pi ^{2} } \right)}_{{CL/F}}\), and was implemented as illustrated for CL/F in Eq. (2):
(CL/F) ij is the oral clearance for individual i on occasion j.
Residual variability, arising from unspecified within-subject variability, model misspecification and experimental error, was described using a slope-intercept model comprising additive (σadd) and constant coefficient of variation (σccv) components.
Model selection was achieved by use of the objective function (OFV), a goodness-of-fit criterion which is minus twice the log likelihood of the data, as well as by evaluation of parameter estimates (including precision) and goodness-of-fit plots. The differences in OFV between a full and a reduced model are approximately chi-square distributed. Differences of ≥10.83 or ≥3.84 (for the FO and FOCE methods, respectively) in the OFV were regarded as significant, corresponding to confidence levels of p<0.001 and p<0.05, respectively. In view of the relatively high levels of type I error observed using the FO method in NONMEM [19], a higher significance level was deemed appropriate in order to compensate for this.
Potential covariate relationships were identified by using stepwise generalized additive modelling (GAM), as implemented in Xpose, and by visual detection of trends in plots of model parameters against covariates. The covariates selected in the GAM analysis were tested in the model by stepwise addition using an OFV change of ≥10.83 (p<0.001) as the cutoff for inclusion, followed by stepwise deletion using an OFV change of ≥12.12 (p<0.0001) as a prerequisite for retention [9].
The true confidence intervals (CIs) of the final model parameters were estimated using log-likelihood profiling (LLP). Census was used to automate the creation and execution of the large number of NONMEM model files required for this technique. Each NONMEM-estimated parameter was independently fixed to −60%, −40%, −20%, −15%, −10%, −5%, 5%, 10%, 15%, 20%, 40% and 60% of its estimated value, leaving all other parameters unchanged, in successive NONMEM runs of the final model. The resulting OFV in each case was plotted against its fixed value to generate a curve for each parameter. Fitting this curve using a 3rd-order polynomial allowed the prediction of parameter values equivalent to a change of 3.84 in the OFV, generating an estimate of the true 95% CI.
Simulations were also used for model evaluation. The final model was used in NONMEM to simulate 1,000 datasets using the original design for the DPM2 and BKH studies. Estimates of area under the concentration-time curve from zero to infinity (\(AUC_{{0 - \infty }}\)) were calculated algebraically from simulated values of CL/F according to Eq. (3).
F in this equation represents oral bioavailability, and Dose is oral PZA dose in mg. \(AUC_{{0 - \infty }}\) was also calculated for observed data in each individual and occasion in the DPM2 and BKH groups by noncompartmental analysis using WinNonlin 4.1 (Pharsight, Mountain View, Calif., USA). The distributions of the simulated and observed (noncompartmental) \(AUC_{{0 - \infty }}\) were compared using a Kolmogorov-Smirnov test (R 2.2.0, R Foundation for Statistical Computing, Vienna, Austria).
Results
The pharmacokinetics of PZA in the studied patients were best described by a one-compartment model with first-order absorption and first-order elimination, parameterized in terms of k a (first-order absorption rate constant), CL/F (apparent oral clearance), V/F (apparent volume of distribution) and duration of zero-order release of the drug from formulation into the absorption compartment (Dur). Exponential IIV terms in CL/F, V/F, and Dur were supported by the data. It was assumed for the purposes of the analysis that bioavailability (F) was equal to 1, since intravenous data for the formulations used was not available. An absorption lag time parameter was not supported by the available data. The final model included IOV in CL/F and k a , as well as covariance between CL/F and V/F. In addition, bimodality in the distribution of k a was characterized by means of a mixture model, which accounted for all the IIV in the parameter. Inclusion of a separate IIV term was not supported by the data. Two subpopulations with significantly different typical values of k a were identified, and will be referred to from this point as fast (k a =3.56 h−1) and slow (k a =1.25 h−1) absorbers. The proportion of fast absorbers in the population was described in the final model by P fast ., and was estimated as 0.556. Final population pharmacokinetic parameter estimates were reasonably precise and are presented in Table 2.
The final model included four covariate relations. Weight was found to increase both CL/F (by 0.545 l h−1 for every 10 kg increase over the population median) and V/F (by 4.33 l for every 10 kg), independently. V/F was also found to be 4.55 l greater in males than females. The use of a fixed-dose combination (FDC) formulation, in which several antimycobacterial drugs are packaged together in the same formulation in fixed dose proportions to aid in treatment adherence, was found to reduce Dur by 5.42 min in the typical individual.
The 95% CIs for CL/F and V/F obtained by the LLP were similar to those calculated from the standard errors (SE) reported by NONMEM, but those obtained from the LLP for the other model parameters were wider and less symmetrical than their SE-derived equivalents. The LLP-generated 95% CIs are listed in Table 2. The observed data is presented as Fig. 1. Goodness-of-fit plots, provided in Figs. 2 and 3, indicated that the final model described the data well. Figure 4 illustrates the validity of the model with respect to \(AUC_{{0 - \infty }}\). There was close similarity between the distribution of values calculated from the observed data and the distribution of simulated values. The distribution of the simulated \(AUC_{{0 - \infty }}\) values was not significantly different from the estimates obtained from noncompartmental analysis (p>0.05).

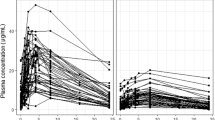
Plot of plasma PZA concentration against time, identifying the different data subgroups (DPM1, DPM2 and BKH)

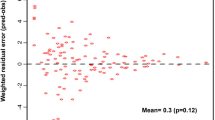
Goodness-of-fit plots for the final model for PZA pharmacokinetics in South African pulmonary tuberculosis patients. DV observed PZA concentration; PRED population predicted PZA concentration; IPRED individual predicted PZA concentration; |IWRES| absolute individual weighted residual; WRES weighted residual

Plots of the observations (open circles), individual predictions (solid lines) and population predictions (dotted lines) from the final model, to illustrate goodness-of-fit for fast absorbers and slow absorbers. Worst fit, Typical fit and Best fit denote degree of goodness-of-fit, categorized by the median of the absolute weighted individual residuals (IWRES) in each individual. C=pyrazinamide plasma concentration

Results of a predictive check, using distributions of values of \(AUC_{{0 - \infty }}\) obtained by simulation using the final PZA model (grey bars, n=1,000) and \(AUC_{{0 - \infty }}\) estimates obtained by noncompartmental analysis of the DPM2 and BKH patient subsets (solid line). The dashed vertical lines represent the median (451 mg h l−1), lower and upper quartiles (381 mg h L−1 and 561 mg h l−1, respectively) of the noncompartmental \(AUC_{{0 - \infty }}\) estimates. The dotted vertical lines represent the median (427 mg h l−1), lower and upper quartiles (346 mg h l−1 and 547 mg h l−1, respectively) for the simulated \(AUC_{{0 - \infty }}\) values
Discussion
The most interesting finding derived from the modelling of PZA pharmacokinetics in the patients studied was the identification of a bimodal distribution in the drug’s rate of absorption. Two discrete subpopulations were found, one in which absorption was rapid, and one in which it was considerably slower. The mixture model incorporated to describe this phenomenon, which accounted for all significant IIV on k a , estimated that 56.6% of the population were fast absorbers. The categorization of a subject as a fast or slow absorber was not related to any studied covariate, although particular care was taken to investigate the effects of formulation, drug intake and study site.
While a bimodal distribution of k a may be assumed to have little clinical relevance, it is interesting to note that a similar bimodal distribution has been detected in RIF, in the same population of patients. In our data, 61% of those characterised as fast absorbers of PZA were fast absorbers of RIF as well (unpublished data, manuscript in preparation). Forty-six percent of the slow absorbers of PZA were also slow absorbers of RIF. In summary, assignment to absorber subgroup, fast or slow, was the same in both drugs in 54% of the 223 patients common to both datasets. This is not far from the results one might expect from a random coin-toss, indicating that the observed bimodality in PZA absorption is probably not mechanistically related to the same phenomenon observed in RIF.
A better fit of the model to the data during the absorption phase was achieved through the inclusion of a parameter describing zero-order release of drug from its oral formulation. Once the drug became available for absorption, it was modelled as subject to a first-order absorption process with a bimodal distribution encapsulating two subpopulations, representing fast and slow absorbers. Dur, the duration of zero-order release of the drug, was quite variable (0.957, variance), but both its typical value and its IIV were well estimated, with percentage relative standard error values (% RSEs) of 1.96% and 25.2%, respectively. Other potential sources of variability in the absorption process include differences in gastrointestinal motility, regional pH changes in the gut, gastric emptying, differences in the effective surface area for absorption, and disease-related factors such as diarrhoea. However, it was not possible to collect data on any of these within the context of what was strictly an observational clinical study.
Some of the variability in Dur was explained by the inclusion of FDC formulation as a covariate. Subjects receiving PZA as part of an FDC were associated with shorter Dur. This suggests that formulation type affects the absorption profile of the drug, probably due to variables such as dissolution and disintegration characteristics. The topic of formulation effects on the bioavailability of tuberculosis drugs, particularly rifampicin, when administered as components of FDC formulations has received considerable attention in the literature recently [1, 14, 15].
Our data supported IOV in k a and CL/F. The latter had similar estimates of IIV (0.0351, variance) and IOV (0.0238, variance). However, inclusion of a parameter describing IIV in k a was not supported by the data. IOV was shared between fast and slow absorbers, and was relatively substantial (0.623, variance). This suggested that some of the variability in the rate of absorption of PZA originated from sources that varied randomly over time and could not be explained by IIV or by the mixture model.
Weight was a significant covariate predictor of CL/F in the final model, which was not unexpected, having been observed before in other settings [3, 24]. According to the final model, a 10 kg increase in weight from the median of 48 kg resulted in an increase of 0.545 l h−1 in CL/F in the typical individual. Weight also predicted V/F, independently of its relationship to CL/F. V/F increased by 0.433 l for every 10 kg increase from the median. In addition, V/F was 4.55 l higher in males compared to females, independently of weight.
The previous population study by Peloquin et al. in North American subjects [12] reported distributions of k a values centred at 3.25±2.36 h−1 and 4.62±5.22 h−1 (mean±SD), using iterative two-stage maximum a posteriori probability Bayesian (IT2B) and nonparametric expectation maximization (NPEM) methods, respectively. The study by Zhu et al. [24], also using NPEM, found median k a values of 1.49 (ranging between 0.40 and 8.87) and 2.73 (ranging between 0.03 and 6.59) in separate groups of 42 adults and 25 adults and 2 children, respectively. These results are broadly comparable to those that we report here, although both previous studies concluded that variability and delays in absorption in adults were small, and no bimodality was noted. Slow absorption of PZA was reported by Zhu et al., but at extremely low frequencies in adults [24]. Population parameter estimates of CL/F and V/F in South African tuberculosis patients in this study were consistent with those reported by Peloquin et al. and by Zhu et al. [12, 24]. No differences with respect to PZA pharmacokinetics between patients and healthy volunteers have been reported in other populations.
As its activity stems almost entirely from its primary metabolite, it would be of interest to investigate the degree to which variability in PZA’s pharmacokinetics influences variability in the pharmacokinetics of POA, as well as pharmacodynamic effects in this population. Without a clear understanding of this relationship, it is difficult to assess the clinical relevance of our findings.
Conclusions
The pharmacokinetics of PZA were described in South African tuberculosis patients. The absorption of PZA in this population was highly variable, and two separate subpopulations with significantly different absorption rates were identified. IOV accounted for a proportion of the variability in clearance and absorption rate constant, with the bivariate mixture model accounting for the variability between patients in the latter. Estimates of oral clearance and volume of distribution were similar to those published in previous reports, and estimates of absorption rate constant fell within the broad ranges reported elsewhere, suggesting that the pharmacokinetics of PZA do not vary substantially between different populations.
References
Agrawal S, Panchagnula R (2005) Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage forms. Biopharm Drug Dispos 26:321–334
Beal SL, Sheiner LB, Boeckmann A (1996) NONMEM Users’ Guides. NONMEM Project Group, University of California, San Francisco
Ellard GA (1969) Absorption, metabolism and excretion of pyrazinamide in man. Tubercle 50:144–158
Hausler H (2000) Tuberculosis & HIV/AIDS: Clinical Guidelines. National Department of Health, South Africa
Heifets L, Lindholm-Levy P (1992) Pyrazinamide sterilizing activity in vitro against semidormant Mycobacterium tuberculosis bacterial populations. Am Rev Respir Dis 145:1223–1225
Jonsson EN, Karlsson MO (1999) Xpose-an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64
Konno K, Feldmann FM, McDermott W (1967) Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis 95:461–469
Maher D, Mikulencak M (1999) What is DOTS? A guide to understanding the WHO-recommended TB control strategy known as DOTS. WHO/CDS/CPC/TB/99.270. World Health Organization, Geneva
Mandema JW, Verotta D, Sheiner LB (1992) Building population pharmacokinetic-pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm 20:511–528
McDermott W, Tompsett R (1954) Activation of pyrazinamide and nicotinamide in acidic environment in vitro. Am Rev Tuberc 70:748–754
Mitchison DA (2000) Role of individual drugs in the chemotherapy of tuberculosis. Int J Tuberc Lung Dis 4:796–806
Peloquin CA, Jaresko GS, Yong CL, Keung AC, Bulpitt AE, Jelliffe RW (1997) Population pharmacokinetic modeling of isoniazid, rifampin, and pyrazinamide. Antimicrob Agents Chemother 41:2670–2679
Salfinger M, Heifets LB (1988) Determination of pyrazinamide MICs for Mycobacterium tuberculosis at different pHs by the radiometric method. Antimicrob Agents Chemother 32:1002–1004
Shishoo CJ, Shah SA, Rathod IS, Savale SS, Vora MJ (2001) Impaired bioavailability of rifampicin in presence of isoniazid from fixed dose combination (FDC) formulation. Int J Pharm 228:53–67
Singh S, Mariappan TT, Sankar R, Sarda N, Singh B (2001) A critical review of the probable reasons for the poor variable bioavailability of rifampicin from anti-tubercular fixed-dose combination (FDC) products, and the likely solutions to the problem. Int J Pharm 228:5–17
Smith PJ, van Dyk J, Fredericks A (1999) Determination of rifampicin, isoniazid and pyrazinamide by high performance liquid chromatography after their simultaneous extraction from plasma. Int J Tuberc Lung Dis 3:S325–S328
Tarshis MS, Weed WA (1953) Lack of significant in vitro sensitivity of Mycobacterium tuberculosis to pyrazinamide on three different solid media. Am Rev Tuberc 67:391–395
The National Essential Drugs List Committee (1998) Standard treatment guidelines and essential drugs list for South Africa: adults: hospital level. National Department of Health, Pretoria
Wählby U, Jonsson EN, Karlsson MO (2001) Assessment of actual significance levels for covariate effects in NONMEM. J Pharmacokinet Pharmacodyn 28:231–252
Wilkins JJ (2005) NONMEMory: a run management tool for NONMEM. Comput Methods Programs Biomed 78:259–267
World Health Organization (2003) Treatment of Tuberculosis: Guidelines for National Programmes. World Health Organization, Geneva
Yeager RL, Munroe WG, Dessau FI (1952) Pyrazinamide (aldinamide) in the treatment of pulmonary tuberculosis. Am Rev Tuberc 65:523–546
Zhang Y, Scorpio A, Nikaido H, Sun Z (1999) Role of acid pH and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J Bacteriol 181:2044–2049
Zhu M, Starke JR, Burman WJ, Steiner P, Stambaugh JJ, Ashkin D, Bulpitt AE, Berning SE, Peloquin CA (2002) Population pharmacokinetic modeling of pyrazinamide in children and adults with tuberculosis. Pharmacotherapy 22:686–695
Acknowledgements
We thank Jean van Dyk, Rudy Onia, Afia Fredericks and Alicia Evans for their invaluable technical and logistical assistance. We also wish to thank Bernard Fourie for allowing the use of some of the data included in this study. This research was co-funded by the South African Medical Research Council and by the Division of Clinical Pharmacology of the Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa. All work described here was carried out with full written ethical approval from the University of Cape Town, Brewelskloof Hospital and the D P Marais SANTA Centre, and complies fully with South African legal requirements for biomedical research.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00228-006-0178-z
Rights and permissions
About this article
Cite this article
Wilkins, J.J., Langdon, G., McIlleron, H. et al. Variability in the population pharmacokinetics of pyrazinamide in South African tuberculosis patients. Eur J Clin Pharmacol 62, 727–735 (2006). https://doi.org/10.1007/s00228-006-0141-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-006-0141-z