
Abstract
Species introductions have been recognized as one of the principal threats to marine environments worldwide. Comparison of genetic data between native and non-native populations can provide key information, such as origin and population demography during the colonization process, which assists in understanding the mechanisms of invasion success in marine environments. The yellowfin goby, Acanthogobius flavimanus, is a large goby native to northeastern Asia, typically inhabiting muddy bottoms of bays, estuaries, and rivers, and is considered a pest where it has invaded coastal areas of the United States and Australia. Here, we analyzed mitochondrial DNA control region sequences of several yellowfin goby populations from both native and non-native distributions. The phylogenetic tree showed no intra-specific lineages, which is in contrast with previous phylogeographic studies that have shown deep genetic divergence in other coastal marine gobies around the Japanese archipelago. On the other hand, at the population level, we found significant genetic differentiation between northern and southern groups in the native distribution, which may be attributed to a rapid population expansion event of the southern group. Our analyses suggest that the origin of the northern California population is Tokyo Bay, but we were unable to identify the original source populations of the southern California and Melbourne populations. These populations showed greatly differing genetic diversities, suggesting their different demographic histories. This study contributes a new perspective on the genetic diversity of multiple populations of the yellowfin goby, as well as representing an example of the relationships between genetic diversity and invasion success.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Species introductions due to canal construction, shipping, and ballast water transport have been recognized as one of the principal threats to marine environments worldwide (Molnar et al. 2008). Evaluating the colonization process during invasion is important to prevent the introduction of new pest species and to assess a species’ relative ecological success in its introduced versus native distributions (Amsellem et al. 2000; Kang et al. 2007). In particular, comparison of genetic data between native and non-native populations can identify dominant haplotypes that have dispersed and persisted in new environments through the colonization process, which enables determination of origins and population demography, and ultimately an understanding of the mechanisms of invasion success (Brown and Stepien 2009; Rius et al. 2015). However, if knowledge of the genetic structure of native distributions is insufficient, then genetic analyses could result in misleading interpretations about the invasion process.
The yellowfin goby, Acanthogobius flavimanus, is a large goby native to northeastern Asia (Japanese archipelago, Primorsky Krai, Korean peninsula, Bohai Sea, and Yellow Sea) (Akihito et al. 2002) that inhabits the muddy bottoms of bays, estuaries, and rivers (Dotsu and Mito 1955). It is exploited both commercially and recreationally in Japan (Shimizu 1984) whereas it is considered a pest species in the coastal areas of the United States (Brittan et al. 1963, 1970; Haaker 1979) and Australia (Middleton 1982). Introduction to these non-native habitats is thought to be through ballast water transport of eggs or larvae (Baltz 1991). Phylogeographic information for this species is limited to one study, where the mitochondrial DNA control region (mtCR) of three native (Tokyo, Fukushima, and Miyagi in the Japanese archipelago) and two invasive (northern and southern California) populations were analyzed (Neilson and Wilson 2005; Fig. 1). They reported genetic similarity among the two Californian populations and the Tokyo population, and low genetic diversity within the southern California population. This suggested that the origins of the two Californian populations were Tokyo Bay and that the southern California population experienced a genetic bottleneck. However, Neilson and Wilson (2005) also suggested that there may be a different origin for the southern California population due to significant genetic differentiation when compared to the investigated native populations. Therefore, a phylogeographic study focusing on a number of populations from wide native distributions was required in order to accurately estimate the invasion processes of this species. In Australia the species is declared a pest, yet prior to this study, there have been no investigations of genetic data from any Australian populations of yellowfin gobies and therefore no capacity to understand the likelihood of it becoming a major pest species, as it has become in some Californian locations.

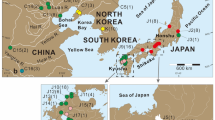
Sampling locations of the yellowfin goby. Details of the locations are shown in Table 1
Here, we analyzed mtCR sequences of several yellowfin goby populations from the Japanese archipelago and Korean peninsula, and an introduced population from Australia. In Australia, yellowfin gobies have been reported along the northern coast of New South Wales and Botany Bay (Sydney), as well as south-eastern Australia including Port Phillip Bay and Western Port, Victoria (Hoese 1973; Bell et al. 1987), although they are not abundant in those locations (personal observations). Our aims were to investigate the phylogeographic structure of native distributions of the yellowfin goby and to attempt to identify source populations involved in the colonization process of the species in non-native populations as well, in order to gain some insights into the likelihood of this species becoming a major marine pest in its non-native distributions.
Materials and methods
Sample collection
A total of 307 fish were sampled for this study (locations shown in Fig. 1; Table 1). 261 samples were collected by angling from 18 native locations on the Japanese and Korean coastlines during 2014–2015. In Australia, 46 samples were collected during 2010–2014 using fyke nets and beach seine nets from four locations around Melbourne, Victoria (Cardinia Creek, Hobsons Bay, Maribyrnong River, and Yarra River). Total DNA was extracted from pectoral fin using Gentra Puregene Tissue Kit (QIAGEN) or a Chelex© extraction method modified from Walsh et al. (1991). Since analyses were conducted in laboratories in both Japan and Australia, there were some minor differences in the methods used to amplify and purify DNA samples; however, these differences were not considered likely to affect the outcomes of the genetic analysis.
Mitochondrial DNA analysis
Partial mtCR of each specimen was amplified using primers provided by Neilson and Wilson (2005) (forward: 5′-TCCCATCTCTAGCTCCCAAA-3′; reverse: 5′-TACGTTGGCGTGTGCATTAT-3′). To improve sequence quality, a slightly modified reverse primer, Rev-2 (5′-GTTGGCGTGTGCATTATTG-3′), was developed and used to amplify and sequence the Australian samples. For amplification, the following reagents were added to each microtube: 100 ng of template DNA, 0.25 U of Tks Gflex DNA polymerase (TAKARA), 5 μL of the 2× PCR reaction buffer, and 0.1 μL of each primer (25 μM). Enough sterile deionized water was added to each microtube to obtain 20 μL. PCR conditions consisted of initial denaturation at 94°C for 4.5 min, followed by 35 cycles of 94°C for 50 s, 55°C for 40 s, and 72°C for 1.5 min, and final extension at 72°C for 5 min. The PCR products were then purified with EXOSAP-IT (USB Corp., Cleveland). All PCR products were sequenced in both directions by Macrogen Inc. (Seoul, South Korea) using an ABI 3730xl capillary sequencer. We additionally downloaded mtCR sequences from the native populations of Tokyo, Fukushima, and Miyagi and non-native populations from northern and southern California deposited in GenBank of National Center for Biotechnology Information (Fig. 1; Table 1). Although Neilson and Wilson (2005) treated the individuals sampled from Bolsa Chica and Newport Bay in southern California as different samples, we combined these samples since their sequences were not significantly different.
Data analysis
Sequences were aligned using the CLC main workbench (QIAGEN) at default settings with manual correction. Although Neilson and Wilson (2005) used the entire mtCR region (986 base pairs), the sequence length in this study was cut down to 711 base pairs due to poor quality of sequencing of some individuals near the primer ends. This processing reduced 80 haplotypes reported by Neilson and Wilson (2005) to 67 haplotypes in this study. Since sequence similarity due to homoplasy can result from saturation of mutations (Bradman et al. 2011), we conducted saturation analysis (Xia et al. 2003) using DAMBE5 (Xia 2013). The index of substation saturation values was much smaller than the critical value, indicating that the sequences are useful in phylogenetic reconstruction (Xia et al. 2003). DNAcollapser of FaBox (Villesen 2007) was used to assign individuals to the haplotype sequences, which were submitted to DNA Data Bank of Japan (DDBJ; LC213730-LC213799; http://www.ddbj.nig.ac.jp). Phylogenetic analyses were carried out on all haplotypes using the maximum-likelihood method in MEGA 5.2.2 (Tamura et al. 2011). MEGA was run for 1000 bootstrap replicates using the T92 + G + I substitution model (Tamura 1992), which was selected as the best-fit model by MEGA, and mtCR sequence of Acanthogobius hasta (Accession Number: AY486321) was used as an outgroup. Phylogenetic relationships among the haplotypes were also estimated by the median-joining and maximum-parsimony methods in NETWORK 5.0.0.0 (Bandelt et al. 1999; Polzin and Daneshmand 2003). BEAST 1.8.0 was used to estimate the time of the common recent ancestor (Drummond and Rambaut 2007). Because a likelihood ratio test did not show rate heterogeneity among the haplotypes of A. flavimanus (P = 0.917), we used the strict clock model with a prior distribution of an evolutionary rate [clock rate = 1.0% ± 1.0%/million year (MY) per lineage] by referring to its typical range in fish (0.4–1.8%/MY per lineage, Tringali et al. 1999; Donaldson and Wilson 1999). In the BEAST analysis, the HKY + G + I substitution model (Hasegawa et al. 1985) and the Yule tree prior were used, where a total of 100 million generations were generated and one in every 1000 generations was sampled in the Markov chain Monte Carlo simulation (10% of the initial samples were removed as burn-in). The convergence of the chains to the stationary distribution and large effective sample size (>300) were confirmed using Tracer 1.5 (Rambaut and Drummond 2009).
Haplotype diversity (h) and nucleotide diversity (π) of each population were calculated using Arlequin 3.5 (Excoffier et al. 2005). The genetic structures in the native and non-native distributions were assessed by hierarchical analysis of molecular variance (AMOVA; Excoffier et al. 1992) and spatial analysis of molecular variance (SAMOVA; Dupanloup et al. 2002). The significance of the genetic differentiation was tested with 1000 permutations of the original dataset. SAMOVA iteratively sought composition of K groups of geographically adjacent populations that maximized ΦCT with 100 simulated annealing processes. Pairwise ΦST was calculated using Arlequin and its significance level was corrected by the Bonferroni method (Rice 1989). A neighbor-joining tree (Saitou and Nei 1987) was constructed based on the net nucleotide differences between the populations, which were calculated using NEIGHBOR in PHYLIP 3.695 (Felsenstein 1993). To test for isolation by distance (Slatkin 1993) in the native distribution, the strength and significance of the relationship between the pairwise values of ΦST/(1 − ΦST) (Rousset 1997) and geographic distances were assessed by the reduced major axis regression and the Mantel test using Isolation by Distance Web Service (Bohonak 2002; Jensen et al. 2005).
Demographic histories of the two groups suggested by SAMOVA were inferred using the mismatch distribution analysis of Arlequin (Rogers and Harpending 1992). In this analysis, we compared the observed frequency distributions of pairwise nucleotide differences with their expected distributions in simulated population expansions, and the goodness-of-fit was evaluated using the estimates of the raggedness statistics and the sum of square deviations. In addition, Tajima’s D test (Tajima 1989) was performed to provide estimates of changes in N e, and its significance was assessed by comparing to the empirical distributions based on 1000 simulated re-samplings under the neutral model. The demographic histories were also inferred using Bayesian skyline plots (BSPs) provided by BEAST. Model parameters consisted of the HKY + G + I substitution model with a strict molecular clock (1.0%/MY per lineage) and a BSP prior, and other settings were at default. BSPs were drawn using Tracer with default settings.
Results
Genetic diversity in the mitochondrial DNA control region
Seventy new haplotypes were obtained from the 261 specimens of A. flavimanus sampled from the native distribution. A total of 137 haplotypes in both the native and non-native populations were used to construct a phylogenetic tree (Fig. 2). The tree revealed no geographical lineages, and this result was unchanged even when we excluded the outgroup to avoid the random rooting effect in phylogenetic reconstruction (Hirase et al. 2016). BEAST analysis estimated that the time of the recent common ancestor was 0.60 million years ago (95% highest posterior density interval: 0.14–1.39). A median-joining network showed a star-like form (Supplementary Figure 1). Haplotype 1 (H1 in Supplementary Fig. 1) was located in its center and contained haplotypes from almost all native and non-native populations.

Maximum-likelihood tree of the 137 yellowfin goby haplotypes with the haplotype frequencies
Genetic structure in native distribution
The haplotype diversity (h) and nucleotide diversity (π) of each of the native populations are summarized in Table 1. The values of h ranged from 0.350 (Mikata) to 1.000 (Fukushima), and those of π ranged from 0.078 (Fukuoka) to 0.616 (Hamamatsu). Haplotype 1 was shared with high frequency (>0.20) in all populations except Otaru, Fukushima, Miyagi, Shimizu, Hamamatsu, and Seikai (Figs. 1, 2). Within the native distribution, AMOVA showed significant genetic variance (ΦST = 0.236, P < 0.001) among the populations. SAMOVA suggested that optimum partitioning was obtained when the populations were divided into two groups: northern (Otaru, Fukushima, and Miyagi) and southern groups (the others). ΦCT reached a plateau at K = 2, whereas several populations contained more than one group at K > 2. This two-group structure was also supported by the population tree (Fig. 3). Significant genetic variance was observed between (ΦCT = 0.414, P < 0.01) and within these two groups (northern: ΦST = 0.087, P < 0.001; southern: ΦST = 0.099, P < 0.001). All except one pairwise ΦST values between the two groups were significant (P < 0.05 after the Bonferroni correction; Table 2) and larger than those within each group.

Unrooted neighbor-joining tree of the 24 yellowfin goby populations based on net nucleotide differences
Only a limited number of populations belonging to the northern group were tested, and therefore the Mantel tests were only performed for the southern group. For the southern group, populations were analyzed separately on the basis of their locations around the Pacific Ocean (Urayasu, Tokyo, Yokohama, Shimizu, Hamamatsu, Handa, Yokkaichi, and Komatsushima) and Sea of Japan coastlines (Niigata, Mikata, Matsue, Shimonoseki, Kitakyushu, Fukuoka, and Seikai), in order to remove effects other than geographic distance (i.e., complex current systems in the Seto Inland Sea and different oceanographic conditions between the Pacific Ocean and Sea of Japan sides; Fig. 1). The Mantel tests showed positive correlations between the genetic and geographic distances in the southern group (along the Pacific Ocean coastlines: r = 0.465, P = 0.033; along the Sea of Japan coastlines: r = 0.692, P = 0.038, Fig. 4).

The relationship between the genetic and geographic distances of the southern group of the yellowfin goby in the native distributions
The shape of the mismatch distributions of the two groups was unimodal and similar to that of simulated distributions under the model of rapid population expansion (Rogers and Harpending 1992); neither the sum of the squared deviation nor the raggedness index suggested significant difference (P > 0.05). Significantly negative Tajima’s D value (−2.20; P = 0.000) supported the rapid population expansion scenario of the southern group but not that of the northern group (−0.81; P = 0.232). Recent and phased increase of the effective population size of the southern group was further supported by BSP analysis (Fig. 5).

Bayesian skyline plot showing changes in N e*t (N e = effective population size; t = generation time) of the southern group of the yellowfin goby in the native distribution. The x-axis is approximate time under the clock rate of 1%/million year per lineage. The y-axis is population size plotted on a logarithmic scale. Black line represents median estimates of the size, whereas the gray area shows the upper and lower 95% highest posterior density range
Genetic structure in non-native distribution
In the non-native populations, haplotype diversity (h) ranged from 0.000 (Melbourne) to 0.918 (northern California), and nucleotide diversity (π) ranged from 0.000 (Melbourne) to 0.359 (northern California) (Table 1). AMOVA showed significant genetic variance (ΦST = 0.107, P < 0.001) among the three non-native populations and all pairwise ΦST were significant (P < 0.05 after Bonferroni correction; Table 3). As in the native populations, Haplotype 1 was most abundant in the non-native populations. In particular, in the Melbourne population, no haplotypes other than Haplotype 1 were found. The population tree including the native and non-native populations showed that the genetic composition of the northern California population was close to those of native populations in Tokyo Bay (Tokyo, Yokohama, and Urayasu; Fig. 3). Furthermore, these four populations shared several haplotypes other than Haplotype 1 (Fig. 2). In contrast, the southern Californian population shared no haplotypes other than Haplotype 1 with any native populations (Fig. 3), and since the Melbourne population had only Haplotype 1, we could not draw conclusions about its origin.
Discussion
Genetic structure of Acanthogobius flavimanus in native distribution
The phylogenetic tree of A. flavimanus showed no intra-specific lineages, which is in contrast with previous phylogeographic studies on other coastal marine gobies that have shown deep genetic divergence (Akihito et al. 2008; Kokita and Nohara 2011; Hirase et al. 2012, 2016; Hirase and Ikeda 2014). A similar comparative phylogeographic pattern in gobiid fish was reported by Dawson et al. (2002), whereby they demonstrated considerably shallower phylogeographic structure in Clevelandia ios compared to Eucyclogobius newberryi, and suggested that this was due to more open habitat, greater abundance, and longer larval duration of C. ios. Like other coastal gobies, A. flavimanus occupies habitat that comprised predominantly open space, and therefore we suggest that life history differences may influence its phylogeographic structure. Firstly, longer pelagic larval durations are expected to result in greater dispersal and lower genetic differentiation (Bohonak 1999; Burton 1983). Indeed, the pelagic larval duration of yellowfin gobies has been estimated to be one month (Suzuki et al. 1989), which is similar to that of C. ios. Secondly, the degree of development in newly hatched larvae may influence phylogeographic structures, because hatching at an advanced stage with superior sensory perception, motor skills, and swimming ability has been suggested to provide an opportunity for larval retention, which could prevent passive dispersal (Blaxter 1986; Brogan 1994). Therefore, if the hatched larvae of A. flavimanus were less developed than other coastal marine gobies, a higher likelihood of passive dispersal could be presumed. However, the sizes of hatched larvae, which can be used as an index to evaluate the degree of development, do not show obvious differences between A. flavimanus (4.6 mm; Suzuki et al. 1989) and other coastal marine gobies [Pterogobius zonoleucus: 4.5 mm; Chaenogobius annularis: 6.1 mm; C. gulosus: 5.5 mm (Japanese Association of Zoos and Aquariums 2007, 2008)]. It may also be noted that P. elapoides and P. zonoleucus have genetic divergence despite a free-floating life history for their entire lifetime. A third consideration of the life history of A. flavimanus that may influence its phylogeographic structure is the use of various environments throughout its life cycle and subsequently an increased susceptibility to environmental changes in these habitats. For example, juveniles settle mainly in shallow estuarine waters such as tidal flats and boulder areas, while adults have been observed to inhabit and reproduce in deeper bay waters (Dotsu and Mito 1955; Sakai et al. 2000; Katayama et al. 2000; Kanou et al. 2005). Changes in the environmental conditions, such as severe oxidation–reduction states in the deep layer in the Sea of Japan during the Pleistocene glacial periods (Itaki et al. 2004) would have likely eliminated some spawning sites in deeper bay waters and thus caused the extinction of ancestral lineages in this region. The contrasting phylogeographic patterns observed within gobiid species around the Japanese archipelago make them an important and interesting group for evaluating the relationships between ecological features and phylogeographic structures, and more studies on other coastal marine gobies are recommended.
While no intra-specific lineages were observed, two genetic groups (northern and southern) were estimated in the native distribution. The time of the recent common ancestor of the observed haplotypes was estimated to be approximately 0.60 million years ago, and the paleoenvironmental changes after this period are expected to be associated with the observed genetic differentiation. Among the two groups, the southern group was characterized by a star-like network, unimodal mismatch distributions, and a significantly negative Tajima’s D value. In addition, we also observed that the frequencies of Haplotype 1, which was at the core of the star-like network, apparently differed between the two groups, with this haplotype observed at high frequency in the southern group. Combined, these results suggest that Haplotype 1 dispersed throughout the southern coastal area around the Japanese archipelago and Korean peninsula more recently, resulting in the genetic differentiation between the northern and southern groups. Bayesian skyline plots (BSP) showed step-wise population expansion of the southern group, and these dates approximately corresponded to the last two interglacial periods (Lisiecki and Raymo 2005). Rapid population expansions during the interglacial periods have been suggested in marine species (Provan et al. 2005; Hoarau et al. 2007; Ni et al. 2014) and coastal marine species in the Japanese archipelago (Kojima et al. 2004; Akihito et al. 2008; Kokita and Nohara 2011; Hirase et al. 2012; Hirase and Ikeda 2014). Therefore, the phased increase of the A. flavimanus population size shown by BSP might reflect the repeated expansion events in the two last interglacial periods. Because isolation by distance was observed in the southern group, it appears that the gene flow between the distant populations was limited after the expansion.
Origin and the genetic diversity of invasive populations
It was suggested that the northern and the southern Californian populations have experienced separate introductions evidenced by the temporal and spatial separation reported in the initial study by Neilson and Wilson (2005). They showed that the haplotypes of the two Californian populations were close to those of Tokyo Bay and distinct from those of Miyagi and Fukushima, suggesting a Tokyo Bay origin. Our study showed that there were two genetic groups in the native distribution, and that Haplotype 1 was detected in high frequency in both groups, as well as the three non-native populations. The population tree showed a close relationship among three native populations sampled in Tokyo Bay and northern Californian populations. Therefore, the origin of the northern Californian population is likely to be Tokyo Bay as suggested in the previous study. The southern Californian population is also likely to be derived from the southern Japanese group, but it was not close to any specific population. There may be two possibilities: the southern California population may be derived from another native population that was not assessed during this study, or it may have undergone a genetic bottleneck (e.g., founder effect and population constriction). Further sampling across both the native and introduced ranges using a multilocus approach would be necessary to fully elucidate the introduction history of the southern California population.
Of the three invasive populations, Melbourne had the lowest genetic diversity with Haplotype 1 being the only haplotype observed. Yet, although the limited samples from Australia are insufficient to thoroughly elucidate the introduction history of the Australian population, a comparison with the native Japanese populations indicates that a genetic bottleneck event may have occurred. This could be attributed to a small number of founders during the colonization process, successive founder effects, or population contraction which may have occurred during colonization due to opposite seasonal changes in sea temperature in the Southern Hemisphere (compared to the Northern Hemisphere), further reducing genetic diversity. A clear need moving forward is the collection of more samples of Australian yellowfin gobies from different populations (i.e., Sydney Harbour and others) to determine if more haplotypes and thus greater genetic diversity is present in other Australian populations of this species.
Genetic bottlenecks are commonly predicted to be associated with invasion (Barrett and Richardson 1986), since invasive populations may be founded by just a few individuals that are isolated from further gene flow (Dlugosch and Parker 2008). Traditionally, the genetic bottleneck has been thought to decrease the potential for adaptive evolution because of the reduction in genetic variation (Frankham et al. 1999). Yellowfin gobies are not common in Australia (Bray and Gomon 2011), which supports our suggestion of a genetic bottleneck in this population, and may prevent further expansion of this species in Australia. Conversely, the high genetic diversity of the northern California population may have led to this species thriving and becoming common in San Francisco Bay (Brittan et al. 1970; Neilson and Wilson 2005), where it has contributed to massive changes in native species interactions and ecosystem structure (Meng et al. 1994). While genetic bottlenecks may restrict the speed of rapid adaptive evolution during most introductions, Prentis et al. (2008) argued that it creates the potential to promote rapid adaptation under special conditions. In the case of Australia, the seasonal change in sea temperatures is remarkably different to those of the native distribution, which is expected to disturb the normal reproduction cycle (Vlaming 1972). Therefore, the genetic bottleneck might have played a crucial role in promoting rapid adaptation, presumably capable of adapting to the vastly different Australian seawater temperatures. In terms of implications, based on the data generated in this study, it would seem that further expansion of the Melbourne population of yellowfin gobies is unlikely, due to low genetic diversity and thus also limited capacity for the adaptive evolution in response to future environmental change. In general, due to repeated introductions, high propagule pressure, and high genetic diversity, invasive populations in marine environments rarely undergo founder effects (Rius et al. 2015). However, our analyses showed greatly differing genetic diversities of the invasive populations, suggesting different demographic histories including a strong founder effect.
In conclusion, this study contributes a new perspective on the genetic diversity of multiple populations of the yellowfin goby, as well as representing an example of the relationships between genetic diversity and invasion success. Combined, this information would offer important insights into some of the factors that drive marine biological invasions.
References
Akihito, Sakamoto K, Ikeda Y, Sugiyama K (2002) Suborder Gobioidei. In: Nakabo T (ed) Fishes of Japan with pictorial keys to the species, English edn. Tokai University Press, Tokyo, pp 1139–1310
Akihito, Fumihito A, Ikeda Y et al (2008) Evolution of Pacific Ocean and the Sea of Japan populations of the gobiid species, Pterogobius elapoides and Pterogobius zonoleucus, based on molecular and morphological analyses. Gene 427:7–18
Amsellem L, Noyer J, Le Bourgeois T et al (2000) Comparison of genetic diversity of the invasive weed Rubus alceifolius Poir. (Rosaceae) in its native range and in areas of introduction, using amplified fragment length polymorphism (AFLP) markers. Mol Ecol 9:443–455
Baltz DM (1991) Introduced fishes in marine systems and inland seas. Biol Conserv 56:151–177
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barrett S, Richardson B (1986) Genetic attributes of invading species. In: Groves RH, Burdon JJ (eds) Ecology of biological invasions. Cambridge University Press, Cambridge, pp 21–33
Bell JD, Steffe AS, Talbot RB (1987) The oriental goby, Acanthogobius flavimanus, colonizes a third estuary in New South Wales, Australia. Ichthyol Res 34:227–230
Blaxter J (1986) Development of sense organs and behaviour of teleost larvae with special reference to feeding and predator avoidance. Trans Am Fish Soc 115:98–114
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Bohonak AJ (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Hered 93:153–154
Bradman H, Grewe P, Appleton B (2011) Direct comparison of mitochondrial markers for the analysis of swordfish population structure. Fish Res 109:95–99
Bray DJ, Gomon MF (2011) Fishes. In: Taxonomic Toolkit for marine life of Port Phillip Bay, Museum Victoria. http://portphillipmarinelife.net.au
Brittan MR, Albrecht AB, Hopkirk JB (1963) An oriental goby collected in the San Joaquin River delta near Stockton, California. Calif Fish Game 49:302–304
Brittan MR, Hopkirk JD, Conners JD et al (1970) Explosive spread of the oriental goby Acanthogobius flavimanus in the San Francisco Bay-Delta region of California. Proc Calif Acad Sci 38:207–214
Brogan MW (1994) Distribution and retention of larval fishes near reefs in the Gulf of California. Mar Ecol Prog Ser 115:1–13
Brown JE, Stepien CA (2009) Invasion genetics of the Eurasian round goby in North America: tracing sources and spread patterns. Mol Ecol 18:64–79
Burton RS (1983) Protein polymorphisms and genetic differentiation of marine invertebrate populations. Mar Biol Lett 4:193–206
Dawson M, Louie K, Barlow M et al (2002) Comparative phylogeography of sympatric sister species, Clevelandia ios and Eucyclogobius newberryi (Teleostei, Gobiidae), across the California Transition Zone. Mol Ecol 11:1065–1075
Dlugosch K, Parker I (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Donaldson KA, Wilson RR Jr (1999) Amphi-panamic geminates of snook (Percoidei: Centropomidae) provide a calibration of the divergence rate in the mitochondrial DNA control region of fishes. Mol Phylogenet Evol 13:208–213
Dotsu Y, Mito S (1955) On the breeding-habits, larvae and young of a goby, Acanthogobius flavimanus (Temminck et Schlegel). Jpn J Ichthyol 4:153–161
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Felsenstein J (1993) Phylogeny inference package (PHYLIP). Version 3.5. University of Washington, Seattle
Frankham R, Lees K, Montgomery ME et al (1999) Do population size bottlenecks reduce evolutionary potential? Anim Conserv 2:255–260
Haaker PL (1979) Two Asiatic gobiid fishes, Tridentiger trigonocephalus and Acanthogobius flavimanus, in southern California. Bull South Calif Acad Sci 78:56–61
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hirase S, Ikeda M (2014) Divergence of mitochondrial DNA lineage of the rocky intertidal goby Chaenogobius gulosus around the Japanese Archipelago: reference to multiple Pleistocene isolation events in the Sea of Japan. Mar Biol 161:565–574
Hirase S, Ikeda M, Kanno M et al (2012) Phylogeography of the intertidal goby Chaenogobius annularis associated with paleoenvironmental changes around the Japanese Archipelago. Mar Ecol Prog Ser 450:167–179
Hirase S, Takeshima H, Nishida M et al (2016) Parallel mitogenome sequencing alleviates random rooting effect in phylogeography. Genome Biol Evol 8:1267–1278
Hoarau G, Coyer J, Veldsink J et al (2007) Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol Ecol 16:3606–3616
Hoese D (1973) The introduction of the gobiid fishes Acanthogobius flavimanus and Tridentiger trigonocephalus into Australia. Koolewong 2:3–5
Itaki T, Ikehara K, Motoyama I et al (2004) Abrupt ventilation changes in the Japan Sea over the last 30 ky: evidence from deep-dwelling radiolarians. Palaeogeogr Palaeoclimatol Palaeoecol 208:263–278
Japanese Association of Zoos and Aquariums (2007) Propagation commendation in fiscal year 2006. J Jpn Assoc Zoos Aquar 48:70 (Japan)
Japanese Association of Zoos and Aquariums (2008) Propagation commendation in fiscal year 2007. J Jpn Assoc Zoos Aquar 49:64–65 (Japan)
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13
Kang M, Buckley YM, Lowe AJ (2007) Testing the role of genetic factors across multiple independent invasions of the shrub Scotch broom (Cytisus scoparius). Mol Ecol 16:4662–4673
Kanou K, Sano M, Kohno H (2005) Ontogenetic diet shift, feeding rhythm, and daily ration of juvenile yellowfin goby Acanthogobius flavimanus on a tidal mudflat in the Tama River estuary, central Japan. Ichthyol Res 52:319–324
Katayama S, Sakai K, Iwata T et al (2000) Life history of Japanese common goby Acanthogobius flavimanus in Hiroura Lagoon of Natori River mouth. Bull Miyagi Pref Fish Res Dev Center 16:93–97 (Japan)
Kojima S, Hayashi I, Kim D et al (2004) Phylogeography of an intertidal direct-developing gastropod Batillaria cumingi around the Japanese Islands. Mar Ecol Prog Ser 276:161–172
Kokita T, Nohara K (2011) Phylogeography and historical demography of the anadromous fish Leucopsarion petersii in relation to geological history and oceanography around the Japanese Archipelago. Mol Ecol 20:143–164
Lisiecki LE, Raymo ME (2005) A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 20:PA1003
Meng L, Moyle PB, Herbold B (1994) Changes in abundance and distribution of native and introduced fishes of Suisun Marsh. Trans Am Fish Soc 123:498–507
Middleton M (1982) The oriental goby, Acanthogobius flavimanus (Temminck and Schlegel), an introduced fish in the coastal waters of New South Wales, Australia. J Fish Biol 21:513–523
Molnar JL, Gamboa RL, Revenga C et al (2008) Assessing the global threat of invasive species to marine biodiversity. Front Ecol Environ 6:485–492
Neilson ME, Wilson RR (2005) mtDNA singletons as evidence of a post-invasion genetic bottleneck in yellowfin goby Acanthogobius flavimanus from San Francisco Bay, California. Mar Ecol Prog Ser 296:197–208
Ni G, Li Q, Kong L et al (2014) Comparative phylogeography in marginal seas of the northwestern Pacific. Mol Ecol 23:534–548
Polzin T, Daneshmand SV (2003) On Steiner trees and minimum spanning trees in hypergraphs. Oper Res Lett 31:12–20
Prentis PJ, Wilson JR, Dormontt EE et al (2008) Adaptive evolution in invasive species. Trends Plant Sci 13:288–294
Provan J, Wattier RA, Maggs CA (2005) Phylogeographic analysis of the red seaweed Palmaria palmata reveals a Pleistocene marine glacial refugium in the English Channel. Mol Ecol 14:793–803
Rambaut A, Drummond A (2009) Tracer version 1.5. 0. WWW document. http://tree.bio.ed.ac.uk/software/tracer/. Accessed 1 Sept 2016
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rius M, Turon X, Bernardi G et al (2015) Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol Invasions 17:869–885
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sakai K, Katayama S, Iwata T (2000) Life history of the Japanese common goby, Acanthogobius flavimanus in the Matsushima Bay. Bull Miyagi Pref Fish Res Dev Center 16:85–92 (Japan)
Shimizu M (1984) Fishes and shellfishes in Tokyo Bay (1). Aquabiology 30:9–13 (Japan)
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Suzuki N, Sakurai N, Sugihara T (1989) Development of eggs, larvae and juveniles of the oriental goby Acanthogobius flavimanus reared in the laboratory. Suisan Zoshoku 364:277–289 (Japan)
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+ C-content biases. Mol Biol Evol 9:678–687
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tringali MD, Bert TM, Seyoum S et al (1999) Molecular phylogenetics and ecological diversification of the transisthmian fish genus Centropomus (Perciformes: Centropomidae). Mol Phylogenet Evol 13:193–207
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7:965–968
Vlaming VL (1972) Environmental control of teleost reproductive cycles: a brief review. J Fish Biol 4:131–140
Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Xia X (2013) DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30:1720–1728
Xia X, Xie Z, Salemi M et al (2003) An index of substitution saturation and its application. Mol Phylogenet Evol 26:1–7
Acknowledgements
The authors thank Kyusyu and Tokushima prefecture lodges of Japan Sport Fishing Foundation, local fishing tackle stores in Japan (Jyosyu-Ya Miyagino store, Point Tokushima store, Anguru Koyaura store, Otaru-fishing PAPA, Kameya-Tsurigu Matsue store), R. Tabata, I. Yokoyama, S. Hayasaka, and T. Mikekado for providing specimens, and S. Matsui, R. Wilson, C. Hayward, M. Lockett, M. McGrouther, and M. Gomon for providing information about yellowfin goby populations. The authors are grateful to the members of the Iwasaki laboratory for helpful comments on this research. This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology (KAKENHI 221S0002 and Project “Construction of the platform for intellectual cooperation”) and the Japan Society for the Promotion of Science (KAKENHI 16H06154 and 26850131). The Australian specimens were collected with support from the Centre for Aquatic Pollution, Identification and Management (CAPIM), Museum Victoria and the Arthur Rylah Institute, Department of Environment, Land, Water and Planning. Additional funding support was received from the Australian Academy of Science (Scientific Visits to Japan, International Linkages Program) and the Australian Society for Fish Biology (Early Career Researcher International Travel Award).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Responsible Editor: T. Reusch.
Reviewed by undisclosed experts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
227_2017_3137_MOESM1_ESM.eps
Supplementary Fig. 1. Median-joining network of the 137 haplotypes of the yellowfin goby. Each line between the haplotypes indicates a single nucleotide substitution. Small black circles between haplotypes represent intermediate hypothesized haplotypes. Circle sizes reflect the sum of the haplotype frequencies of all locations (EPS 995 kb)
Rights and permissions
About this article

Cite this article
Hirase, S., Chambers, S., Hassell, K. et al. Phylogeography of the yellowfin goby Acanthogobius flavimanus in native and non-native distributions. Mar Biol 164, 106 (2017). https://doi.org/10.1007/s00227-017-3137-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-017-3137-6