
Abstract
Ca-phosphate/hydroxyapatite (HA) crystals constitute the mineral matrix of vertebrate bones, while Ca-carbonate is the predominant mineral of many invertebrates, like mollusks. Recent results suggest that CaCO3 is also synthesized during early bone formation. We demonstrate that carbonic anhydrase-driven CaCO3 formation in vitro is activated by organic extracts from the demosponge Suberites domuncula as well as by quinolinic acid, one component isolated from these extracts. Further results revealed that the stimulatory effect of bicarbonate (HCO3 −) ions on mineralization of osteoblast-like SaOS-2 cells is strongly enhanced if the cells are exposed to inorganic polyphosphate (polyP), a linear polymer of phosphate linked by energy-rich phosphodiester bonds. The effect of polyP, administered as polyP (Ca2+ salt), on HA formation was found to be amplified by addition of the carbonic anhydrase-activating sponge extract or quinolinic acid. Our results support the assumption that CaCO3 deposits, acting as bio-seeds for Ca-carbonated phosphate formation, are formed as an intermediate during HA mineralization and that the carbonic anhydrase-mediated formation of those deposits is under a positive–negative feedback control by bone alkaline phosphatase-dependent polyP metabolism, offering new targets for therapy of bone diseases/defects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bone, as a biomineral, is composed of an organic matrix (mainly collagen, ≈25 % w/w), a mineral phase (Ca deposits, ≈65 % w/w), and 10 % water [1, 2]. The dominant component of the inorganic scaffold is calcium phosphate, more specifically calcium hydroxyapatite (Ca–HA, Ca5[PO4]3[OH]). However, bone mineral contains, in addition, around 5 % w/w carbonate, such as carbonate-fluorapatite (francolite) [3]. Earlier studies suggested that carbonate exists in the apatite crystal lattices as CO3 2− ions, perhaps substituting for PO4 3− and/or OH− ions [reviewed in 4]. Topographically, carbonate has been localized in vivo, even as crystalline calcium carbonate (CaCO3) within medullary bone [5]. It has been determined that the bone CO2 compartment consists of 30 % bicarbonate (HCO3 −) and 70 % carbonate (CO3 2−) [6]. In humans/mammals, besides Ca-phosphate-based skeletal elements, biomineral deposits predominantly formed of CaCO3 exist, e.g., the biomineralized otoliths in the vestibular labyrinth of the ear [7, 8]. In otoliths an organic matrix protein, covered with 90–95 % of CaCO3, exists that has been identified as otolin. In addition, otoconins, calcium-binding proteins, exist in the vestibular system, onto which CaCO3 deposition is initiated [9].
Based on the observation that in humans, as a result of metabolic acidosis occurring during aging, a counteraction to the retained endogenous acids proceeds that might result in anabolic processes from the skeleton, followed by a decrease in bone mass; thus, the diet of postmenopausal women was supplemented with potassium bicarbonate. The results revealed that those subject groups show improved calcium and phosphorus balance and reduced bone resorption, resulting in an increased rate of bone formation [10]. Already, bicarbonate supplementation in the diet had been described to improve the nutritional status of patients with chronic kidney disease [11].
Both CaCO3 and Ca-phosphate have been described to precipitate under physiological conditions [12]. Already in this early study it was reported that CaCO3 precipitation was prevented in the presence of low phosphate concentrations, and in turn, it has been suggested that in the biological system metabolic processes that locally raise the bicarbonate level and simultaneously lower the phosphate concentration are required for mineral deposition. HCO3 − accumulates on the surfaces of cell membranes in the vicinity of the Na+-driven Cl−/HCO3 − exchanger (NBC) [13], the Na+–HCO3 − cotransporter [14], and the Na+-independent Cl−/HCO3 − exchanger (AE) [15], all of them being (primarily) involved in the regulation of intracellular pH. Interestingly, some of those channels are connected with carbonic anhydrase (CA), like the AE3 Cl−/HCO3 − exchanger and CA XIV [16]. CA enzymes catalyze the rapid interconversion of one carbon dioxide molecule and water to HCO3 − and one proton. Under physiological conditions, the CA-driven HCO3 − formation is the rate-limiting reaction during carbonate deposition in animals, e.g., in mollusk shells [17]. Among mammalian CAs the most abundant isoforms are the cytosolic CA II, with about 75 %, and the cytosolic CA I, with about 20 % [18]. One considerable fraction of those CAs is cell membrane-associated, both for CA II [19] and for other isoforms [20].
Recently, more detailed data on the initiation of mineralization in osteoblasts, the bone-forming cells, have been elaborated. Ca deposition in osteoblasts starts intracellularly in calcium-containing vesicles under the formation of poorly crystalline carbonated apatite deposits [21]. Evidence has been presented showing that amorphous Ca-phosphate particles are formed intracellularly using phosphate formed from polyphosphate (polyp) after hydrolytic degradation of this polymer [22], a process that is under enzymatic control. On the other hand, it has been proposed that the extracellular fluid is sufficiently saturated with respect to Ca2+ and phosphate, allowing Ca-phosphate to precipitate [23]. As for (almost) any kind of biological reaction that proceeds within a living biological system, enzymes are involved that allow also exergonic reaction to run under the formation of a chemical bond. In addition, in contrast to chemical reactions, which are usually performed under saturating conditions, biological/enzymatic processes proceed under unsaturated substrate conditions, where a subtle regulation of reaction velocity, by slight changes of the substrate, can be obtained. A first example that even inorganic biological skeletal elements, the siliceous spicules from sponges, are formed enzymatically is silicatein, the major proteinaceous component of spicules [reviewed in 24, 25]. Surely, not only enzymes but also interfaces between two phases can lower the activation energy, allowing exergonic reactions to occur, e.g., in atomic/molecular transport processes [26].
CAs have been identified not only in the anabolic bone-forming osteoblasts but also, though at higher levels, in the catabolic bone-resorbing cells, the osteoclasts [27]. CA II deficiency in humans is a rare autosomal recessive disorder that is characterized by the triad of osteopetrosis, renal tubular acidosis, and cerebral calcifications [28]. In view of this fact it came as a surprise that in vivo administration of CA inhibitors does not affect the function of osteoclasts [29]. Only little information is available about the potential therapeutic effect of CA inhibitors on bone anabolism [30]. Until now only a few CA activators have been identified, but none has been tested for its potential in the treatment of bone disorders [reviewed in 31–33]. This view has changed to some extent by the findings that (1) CaCO3 deposits might function as bio-seeds for Ca-phosphate precipitation onto bone-forming cells [34] and (2) the CAs are decisively involved in CaCO3 deposition in vitro [35] and likely in vivo [36]. The latter studies were performed with the calcareous sponge Sycon raphanus, which has a calcareous CaCO3-based skeleton. Because all metazoan phyla, including the oldest animals, the sponges (phylum Porifera), originate from one ancestor, the Urmetazoa [37], it was tempting to speculate that even the inorganic skeletons of the more evolved animals, e.g., Ca-phosphate-based skeletons, might have components and structures in common with this basal animal taxon.
It is well established that mineralization of osteoblasts, bone mineral-forming cells, is significantly induced by polyP in vitro even in the presence of orthophosphate [38]. In addition, polyP turned out to be an inducer of osteoblast-specific alkaline phosphatase. This finding is interesting in view of published data indicating that intracellularly polyP might be formed in the vesicles of bone-forming cells as Ca2+ salt, which is supposed to act as a potential precursor of carbonated HA [39].
In the present study we show that CA-driven CaCO3 deposition can be stimulated by CA activators both in vitro and in intact cell system (SaOS-2). As activator(s) we chose extracts from the sponge Suberites domuncula and one component, isolated from these extracts, quinolinic acid (QA). This effect is amplified by polyP. Mesenchymal stem cells, which differentiate to osteoblasts, are known to release a factor that attenuates QA toxicity [40]. The rationale behind this study was the finding that even siliceous demosponges form CaCO3 deposits [41], suggesting that also the siliceous sponges might be provided with both anabolic and catabolic biocalcite-forming enzymes and inhibitors of them.
Materials and Methods
Cultivation of SaOS-2 Cells
SaOS-2 cells (human osteogenic sarcoma cells [42]) were cultured in McCoy’s medium (Biochrom-Seromed, Berlin, Germany), lacking Na-bicarbonate but containing 2 mM l-glutamine and 1 mM CaCl2 [43]. The medium was supplemented with 10 % heat-inactivated fetal calf serum (FCS) and 100 units mL−1 penicillin/100 μg mL−1 streptomycin. Where indicated 20 mM Na-bicarbonate (NaHCO3) was added as well. The medium/serum was buffered with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) to a pH of 7.4. The cells were incubated in 25-cm2 flasks or in six-well plates (surface area 9.46 cm2; Orange Scientifique, Braine-l’Alleud, Belgium) in a humidified incubator at 37 °C [44, 45]. Routinely, 3 × 105 cells/well were added (total volume 3 mL). Where indicated, cultures were supplemented with a mineralization activation cocktail (MAC), composed of 5 mM β-glycerophosphate, 50 mM ascorbic acid, and 10 nM dexamethasone [46]. The MAC was added 3 days after starting the experiments. Medium was changed every 3 days, and new MAC was added.
Where indicated with the respective assay, either S. domuncula extract (SubDo-extr) or QA was added at the given concentration.
Mineralization by SaOS-2 Cells In Vitro
The extent of mineralization in assays with SaOS-2 cells was determined quantitatively using the alizarin red S spectrophotometric assay [47, 48]. The amount of bound alizarin red S is given in nanomoles. Values were normalized to total DNA in the samples.
In one series of experiments the cultures, growing in culture wells, were stained directly on the coverslips with 10 % alizarin red S, after fixation with ethanol [44].
Incubation of SaOS-2 Cells with PolyP
SaOS-2 cells were incubated, under otherwise identical conditions, with polyP. As source, Na-polyP (average chain of approximately 40 phosphate units), obtained from Chemische Fabrik Budenheim (Budenheim, Germany), was used for the studies. To compensate for any effect caused by the chelating activity to Ca2+ of polyP after hydrolysis of the polymer to monomeric phosphate by phosphatases in vitro, polyP was mixed with CaCl2 in a stoichiometric ratio of 2:1 (polyP:CaCl2), as described [38]. The salt, designated as polyP (Ca2+ salt), was added at a concentration of 50 μM to the assays.
CA-Mediated CaCO3 Precipitation Assay
For the CaCO3 precipitation assay the ammonium carbonate diffusion method was used, as described [36]. In a 3.4-L large desiccator CO2 vapor was generated from a 1-M NH4HCO3 (A6141; Sigma, Taufkirchen, Germany) solution, which had been placed into the lower compartment of the desiccator. CO2 diffused into the upper compartment, which contained in eight-well square chamber slides 50 mM CaCl2 in 30 mM Tris-HCl (pH 8.3). The slides with an edge length of 10 mm (Lab-Tek II slides 154534) came from Thermo Scientific (Dreieich, Germany). The surface area of the 500-μL assay was 0.7 cm2. Where indicated, the recombinant human CA2 enzyme, expressed in Escherichia coli (C6624, Sigma), with a specific activity of ≈5,000 units/mg [49, 50], was added at a concentration of 35 W-A units (10 μg)/500 μL of CaCl2 to the assays. Prior to the addition of the Tris/CaCl2 reaction vessels, the desiccator was preequilibrated with CO2. The formation of CaCO3 was quantitatively determined on the basis of the consumption of free Ca2+ using the EDTA titration procedure [36, 51].
Crystals were visualized using a light digital microscope (VHX-600 Digital Microscope) from KEYENCE (Neu-Isenburg, Germany).
Preparation of S. domuncula Extract
Freshly collected sponge specimens, dredged 5 km offshore from Rovinj (Adriatic Sea, Croatia), were cleaned and cut into small pieces. The material (1 kg) was homogenized in a Waring blender in the presence of 100 mL of ethanol; then, the slurry was extracted with ethyl acetate (3 × 250 mL) at room temperature. The combined ethyl acetate extracts were washed with water (3 × 200 mL), dried over Mg2SO4, filtered, and evaporated to dryness, yielding ≈4 g of dark tar-like residue. This material, termed SubDo-extr, was dissolved in dimethyl sulfoxide (DMSO) and subjected to testing in the cell culture. Control cultures received the carrier solvent alone (1.0 % DMSO).
Extraction and Identification of QA
QA was extracted as described [52]. The extract was shaken in a separation funnel with chloroform to eliminate nonpolar components. The residue was used for the determination of QA by high-performance liquid chromatography. The eluate was monitored with ultraviolet detection at 268 nm. At this wavelength the molar extinction coefficient (ε) is 4,000 M−1 cm−1 [53].
Scanning Electron Microscopy and Energy-Dispersive X-Ray Analysis
Scanning electron microscopic (SEM) analysis was performed with an SU 8000 (Hitachi High-Technologies Europe, Krefeld, Germany) microscope, employed at low voltage (<1 kV, analysis of near-surface organic surfaces) [38]. The SEM was coupled to an XFlash 5010 detector, an X-ray detector that allows simultaneous energy-dispersive X-ray (EDX)-based elemental analyses. Likewise, the same combination of devices was used for higher-voltage (10 kV) analysis, during which the XFlash 5010 detector was used for element mapping of the surfaces of the deposits. The HyperMap database was used for interpretation, as described [54].
Additional Methods
The results were statistically evaluated [55]. DNA content was determined by application of the PicoGreen method, as described [44], using calf thymus DNA as a standard.
Results
Effect of Sponge Extracts and Sponge QA on CA-Mediated CaCO3 Deposit Formation
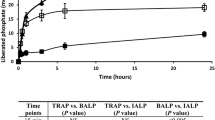
Under the conditions described in “Materials and Methods,” 20 % of the soluble CaCl2 salt was converted to insoluble CaCO3 after an incubation period of 240 minutes using the ammonium carbonate diffusion assay, in the absence of CA (Fig. 1a). Addition of 10 μg × mL−1 of SubDo-extr or of 10 μM QA did not significantly change the extent of CaCO3 formation; the amount of CaCO3 formation was 32.5 ± 4.9 % in the presence of SubDo-extr and 28.4 ± 3.7 % in the presence of QA after the 240-minute incubation period.

Formation of CaCO3 in the ammonium carbonate diffusion assay in the absence a or presence b of 35 W-A units CA per 500-μL crystallization assay. The reaction assays either remained free of additional compound(s) (filled square) or were supplemented with 10 μg × mL−1 of S. domuncula extract (SubDo-extr, filled circle) or with 10 μM quinolinic acid (QA, filled triangle). At the indicated time points samples were taken and the free Ca2+ concentration was determined. The decrease in the concentration of free Ca2+ indicates an increase in deposited CaCO3. Samples of six parallel determinations were quantitated; means ± SD are given. *p < 0.05
In contrast, in the presence of CA the rate of CaCO3 formation increased significantly to 38.4 ± 5.2 % over 240 minutes. Even more, if CA was administered together with QA, a significant increase of mineralization was seen, which amounts to 69.2 ± 7.8 %. In the presence of SubDo-extr, the increase was 83.1 ± 8.1 % (Fig. 1b).
Rate of CaCO3 Crystal Formation Depending on the Presence of CA, SubDo-extr, and QA
No crystals were seen at the beginning of the reaction in the diffusion/desiccator assays (Fig. 2a, b). In the absence of CA, only very scarce, 42 ± 3 μm (n = 10), large crystals were seen after 30 minutes (Fig. 2c); in contrast, in the presence of CA 105 ± 16 μm-sized crystals were regularly seen (Fig. 2d). After an incubation period of 60 minutes, the size of the crystals increased to 122 ± 25 μm in both assays (Fig. 2e, f), again with a much higher abundance in the assays with CA. After a further, extended incubation for 180 min (Fig. 2g, h), the crystals, especially in the assays containing CA, further increased in size. Especially in the assays containing CA two forms of calcitic crystals were seen: (1) round-shaped deposits and (2) rhombohedral prisms. The round-shaped deposits consisted of the metastable vaterite with the characteristic peaks of FTIR spectra at 875 and 744 cm−1, while the prisms consisted of calcite with FTIR peaks at 873 and 711 cm−1, as determined by us previously [35].

Intensified calcitic crystal formation in the carbonate diffusion assay in the presence of 35 W-A units CA/500 μL (b, d, f, h) compared to assays without the enzyme (a, c, e, g). Two different morphologies of the calcitic crystals can be distinguished, the round-shaped vaterite deposits (d) and the rhombohedral prisms of calcite (p). Light microscopic images
In the presence of SubDo-extr, 10 μg × mL−1, the abundance of the calcitic crystals increased significantly in the assays. While at time zero again no crystals were seen (Fig. 3a, b), the number of crystals increased gradually during incubation from 30 minutes (Fig. 3c, d), over 60 min (Fig. 3e, f), to 180 minutes (Fig. 3g, h). The number of crystals in the CA-containing assays significantly higher (Fig. 3d, f, h) compared to the number in those lacking the enzyme (Fig. 3c, e, g).

Crystallization studies in the carbonate diffusion assay were performed again in the absence (a, c, e, and g) or presence (b, d, f, and h) of 35 W-A units/500 μL of CA. All assays contained, in addition, 10 μg × mL−1 S. domuncula extract. Some vaterite deposits (d) and rhombohedral prisms of calcite (p) are marked
A similar effect on the number of crystals formed can be seen if the crystallization studies are performed with 10 μM QA. Again, the prevalence of the distinctly formed crystals increased during the 180-minute incubation period (Fig. 4), especially in the assays containing the CA (Fig. 4d, f, h) compared to the assays lacking CA (Fig. 4c, e, g). However, in addition to the crystals, small-sized deposits were seen together with the crystals that accumulate on the aqueous/air interphase.

In vitro crystallization assay in the absence (a, c, e, g) or presence (b, d, f, h) of 35 W-A units/500 μL of CA; assays were supplemented with 10 μM quinolinic acid. Some vaterite deposits (d) and rhombohedral prisms of calcite (p) are marked
Effect of PolyP and CaCO3 on Mineralization of SaOS-2 Cells
As expected, addition of MAC to SaOS-2 cells caused an increase in mineralization, which became significant after an incubation period of 3 days. The extent of the MAC-caused increase in mineral deposition was lower in the absence of HCO3 − in the assays (Fig. 5a) compared with the increase measured in the presence of HCO3 − (Fig. 5b). At day 5 the ratio/increase of mineral deposition between differentiated (+MAC) and undifferentiated (−MAC) cells was significantly higher in the presence of HCO3 − (Fig. 5b, 5.3-fold increase) compared to the same ratio in the absence of HCO3 − (Fig. 5a, 1.3-fold increase). In contrast, if polyP (Ca2+ salt) was added to the cultures, the MAC-caused stimulation, e.g., at day 5, in the presence of polyP (Ca2+ salt) was significantly higher in the presence of HCO3 − (a calculated 3.6-fold increased ratio, Fig. 5b) compared to experiments in the absence of HCO3 − (1.5-fold, Fig. 5a). Even more, the polyP (Ca2+ salt)-mediated stimulatory effect was even a potentiating one: the increase caused by MAC was 4.8-fold and that by polyP (Ca2+ salt) in the presence of MAC was 17.6-fold, with respect to the effect seen in the absence of MAC. As measured, the basal mineral deposition in the controls (without MAC), cultured in the presence of HCO3 −, resulted in a decrease during the 5-day incubation period. At present, we attribute this finding to a dissolution of the initial amorphous Ca-carbonate, which is highly soluble [56].

Effect of polyP (Ca2+ salt) on the extent of mineralization of SaOS-2 cells in the absence a or presence b of HCO3 −; alizarin red S spectrophotometric assay. As indicated, experiments were performed in the absence (−MAC, open bars/left hatched in black) or presence (+MAC, gray-colored bars/right hatched in white) of MAC. As marked, polyP (Ca2+ salt) was added in the absence of MAC (−MAC + polyP, left hatched in black) or presence (+MAC + polyP, right hatched in white) of MAC. Standard errors of the means are shown (n = 6); *p < 0.05. MAC mineralization activation cocktail, polyP polyphosphate, AR alizarin red
Effect of SubDo-extr and QA on Biomineralization of SaOS-2 Cells
In assays supplemented with 20 mM NaHCO3, both SubDo-extr and QA did not significantly change the low extent of mineralization in the absence of MAC, ≈0.1 nmol alizarin red S staining/1 μg DNA at days 1 and 3, or in medium lacking HCO3 −, ≈0.1 nmol/μg (data not shown). In the presence of MAC, SubDo-extr and QA caused only a small increase in mineralization of SaOS-2 cells, as demonstrated by staining with alizarin red S (Fig. 6). However, in the presence of both MAC and polyP (Ca2+ salt) the two natural components, SubDo-extr and QA, significantly increased the extent of mineral deposition onto SaOS-2, as determined by the quantitative alizarin red spectrophotometric assay (Fig. 7). This increase was especially pronounced at day 3; at this time point the extent of biomineralization, after addition of 5 μg SubDo-extr or of 10 μM QA per assay, was amplified to 2.3-fold or 2.1-fold, respectively. At day 5 of mineralization, the increase was still significant but less pronounced.

Alizarin red staining of cells incubated in medium supplemented with HCO3 − in either the absence (–MAC) or the presence (+MAC) of MAC in dependence on the S. domuncula compounds. Assays were incubated without the sponge compound(s) (−QA/−SubDo-extr) or with the compounds (+QA/+SubDo-extr). The incubation was terminated after 5 days, and coverslips were stained with 10 % alizarin red S. MAC mineralization activation cocktail, QA quinolinic acid, SubDo-extr S. domuncula extract

Stimulatory effect of coincubation of SaOS-2 cells with MAC and polyP (Ca2+ salt) in medium supplemented with HCO3 − (BiCa), if administered together with 10 μM QA (stippled bars) or 5 μg per assay of SubDo-extr (filled bars). Control values without QA or SubDo-extr are given in comparison (right hatched in white). Means (n = 6) with *p < 0.05 are given. MAC mineralization activation cocktail, QA quinolinic acid, SubDo-extr S. domuncula extract, AR alizarin red, polyP, polyphosphate, BiCa bicarbonate
Carbon-Rich Deposits Formed onto SaOS-2 Cells after HCO3 − Exposure: Element Mapping
The cells were incubated in the presence of HCO3 − and MAC for 5 days and then, after fixation, subjected to element mapping, using two-dimensional element mapping in the SEM/EDX setup adjustment (Fig. 8). As shown in Fig. 8a, the nodules are highlighted at the secondary electron image setting. Mapping studies revealed that within the crystallite nodules a strong increase of the impulses for Ca (Fig. 8b), P (Fig. 8c), and C (Fig. 8d) lights up.

Semiquantitative determinations of the accumulation of calcium (Ca), phosphorus (P), and carbon (C) around growing nodules (no), which have been distinguished by secondary electron imaging (SEI). Element mapping was performed by scanning electron microscopy-based energy-dispersive X-ray mapping. a Secondary electron image. Mapping for Ca (b), P (c), and C (d). The intensity scale for the pseudocolor documentation is given below the images; dark/blue low intensities, red/white high intensities. SaOS-2 cells were incubated in the presence of HCO3 − and mineralization activation cocktail for 5 days (Color figure online)
The same series of experiments were performed for SaOS-2 cells that had been grown in the presence of HCO3 − and MAC, again for 5 days, if to this experimental setup 10 μM QA is added as well (Fig. 9). The growing nodules can be distinguished by secondary electron imaging (Fig. 9a). Within the areas of nodules a strong increase of the impulses for Ca (Fig. 9b), P (Fig. 9c), and especially C (Fig. 9d) is seen. It should be particularly mentioned that the regions with increased C signals expand those regions highlighting the nodules (secondary electron imaging) and Ca or P.

Mapping studies for the elements calcium (Ca), phosphorus (P), and carbon (C) around growing nodules (no) onto SaOS-2 cells. Cells were grown for 5 days in the presence of HCO3 − and mineralization activation cocktail, as well as 10 μM quinolinic acid. a Secondary electron image. Mapping for Ca (b), P (c), and C (d). It is obvious that the areas of increased impulses for C (d) extend the areas of increased signals for Ca (b) and P (c)
Discussion
The biomineralization process in osteoblasts starts with the intracellular accumulation of the respective mineralizing ions, HCO3 −, CO3 2−, PO4 3−, and Ca2+, most likely in vesicles, from where they are sequestered to the sites where mineral deposition occurs. Basically, three mechanisms have been proposed to explain early bone mineral formation [21]: (1) a cell-independent process, which suggests that the ions are deposited extracellularly onto charged noncollagenous proteins; (2) a cell-controlled mechanism, via which the intracellular vesicles bud from the plasma membrane and subsequently form Ca-phosphate deposits; and (3) already intracellularly the first amorphous Ca deposits are formed, which are deposited in a further step onto collagen fibrils, where they are transformed to more crystalline apatite platelets. Recent studies favor the last proposal, implying that amorphous mineral precursors act as bio-seeds for bone mineralization [57] followed by a crystallization process [58].
The process of bone mineral formation proceeds on the surface of the bone-forming cells, either in close spatial connection with collagen [59] or on other distinct nucleating sites on the cells [60]. In the latter view it is proposed that bio-seed formation/nucleation starts with the formation of calcium phosphate aggregates that subsequently become densified. After a transient generation of an amorphous precursor phase, the deposits might transform into the crystalline apatite. However, if the biomimetic process is performed under physiological conditions, the mineral deposits are formed of oriented nanocrystallites of carbonated HA. In the amorphous, nanostructured state a stoichiometric replacement of the ionic substitutions can occur, e.g., CO3 2− replacing PO4 3− and Mg2+ replacing Ca2+ [61, 62]. Based on chemical and spectroscopic analyses it has been proposed that bone formation starts from collagen fibrils around which poorly crystalline carbonated apatite crystals, carbonate apatite aggregates, are deposited [21, 63]. These results revealed that under in vitro conditions osteoblasts contain low concentrations of CO3 2− ions within their Ca2+/PO4 3− mineral phase.
As intracellular precursor for Ca-phosphate formation, PO4 3−, deposited as polyP polymer, has been proposed [39]. This polymer has previously been shown to act anabolically on Ca-phosphate formation in SaOS-2 cells in vitro [38, 64]. Even more, this polymer acts as an inducer for the expression of the osteoblast-specific alkaline phosphatase [38]. The experiments suggested that polyP is deposited in vesicles as insoluble Ca2+-polyP salt [39].
Since both Ca-phosphate formation and CaCO3 formation are exergonic reactions and, in analogy to (most) metabolic reactions proceeding in living systems, run at nonsaturating conditions, we postulated that mineralization processes of bone material are also enzyme-driven. Focusing on CaCO3 CA is assumed to be the prime enzyme involved in CaCO3 deposition. Recently, we described that HCO3 − causes an induction of CA in SaOS-2 cells and a subsequent increase in mineralization [34]. Therefore, we screened for CA activators and used sponges as a biological starting material. Control of CA in these animals is crucial for the maintenance of their intermediate metabolism as well as their survival since they live in an environment that is poor in CO2 [65]. While an intensive and successful search for CA inhibitors has been launched, only comparably minor activities have been performed to screen for CA activators [32]. We screened organic extracts from sponges, and among them the ethyl-acetate extract from the demosponge S. domuncula showed a distinct activation of the CA enzyme during in vitro CaCO3 formation. In the presence of 10 μg mL−1 a severalfold increase of the transformation of free Ca2+ into insoluble CaCO3 was found. No distinct differences in the crystal form and state could be detected. During the 240-minute incubation period the enzymatically formed CaCO3 deposits exist in the calcite form, based on the FTIR spectra. Functionally important was the finding that in the presence of HCO3 − and polyP SubDo-extr caused a significant increase in the extent of mineralization in the in vitro assay to 232 % compared to the controls (100 %).
Among the secondary metabolites isolated from S. domuncula [66, 67], we selected QA since this S. domuncula bioactive compound [52] comprises, with its N-heteroatom in the pyridine backbone as well as the dicarboxylic acid side chains, two potential interacting groups with the Zn-containing CA. The zinc prosthetic group in the CA is coordinated in three positions by histidine side chains, by His94, His96, and His119 [68]. The fourth coordination position at Zn is occupied by one water molecule and causes there a polarization of the hydrogen–oxygen bond, resulting in an increased negativity of the oxygen and a weakening of the bond to the Zn atom. A fourth histidine, His64, is placed close to the substrate of water and accepts a proton from the water molecule through general acid-base catalysis under formation of a hydroxide bound to the zinc (Fig. 10). These studies have been performed with human crystallized CA. In analogy to amino acid derivatives [69], the formation of a hydrogen bridge bond can be postulated between the N-heteroatom of QA and the water130 that facilitates the rate-limiting proton transfer step. The dicarboxylic acid side chains of QA could interact, via their conjugated side chains, with the enzyme-bound Zn ion, like in the bipyridyl Zn chelators [70] (Fig. 10). In the future, we plan to modify the position of the carboxylic acid side chain at the pyridine backbone to modulate the basic heterocyclic organic compound through an induction of the delocalization of the π electron within the ring structure.

Proposed interaction of quinolinic acid (QA, in red) with the active center of the human carbonic anhydrase. Three His residues are interacting with the central Zn2+ ion. The three-hydrogen bond network, formed by Wat-150, Wat-129, and Wat-130, is indicated [modified after 75]. It is proposed that QA interacts with its N-heteroatom to Wat-130 and its one carboxylic acid group to Zn2+ (Color figure online)
The mineralization studies with alizarin red also revealed that QA causes a significant upregulation of the biomineralization process by SaOS-2 cells in vitro. This effect is especially pronounced during the initial phase of mineralization, during the first 3 days. At this time point the increase caused by QA is 210 % compared to the 100 % of the controls. Studies using SEM coupled with EDX analysis confirmed our previous findings [34] that carbon is strongly accumulated in the mineral nodules on the surface of the SaOS-2 cells, whether incubated with SubDo-extr or with QA.
The experimental data given here show that at the concentration of 10 μM (0.85 μg/500 μL assay) chosen, QA causes a similarly strong activation compared to the 10 μg × mL−1 of SubDo-extr. This concentration of QA is very low and does not cause apoptotic signs to neuronal cells in vitro [71]. The higher efficacy of QA, on the basis of weight, indicates that it is one of the major constituents of the extract, activating the CA.
As outlined, polyP, likely as polyP (Ca2+ salt), is stored in bone cells [39]. Released from the vesicles, polyP undergoes enzymatic degradation into Ca2+ and PO4 3−, two precursors required for Ca-phosphate formation in SaOS-2 cells [38], via alkaline phosphatase [64]. In a second approach the effect of polyP on the mineralization of SaOS-2 cells in the absence and presence of HCO3 − was tested. We demonstrated for the first time that an enhancement of mineralization of polyP (Ca2+ salt) by HCO3 − in the presence of MAC occurs. This finding is remarkable since polyP causes this effect in the presence of orthophosphate (Fig. 11), present in the culture medium. This finding implies that polyP, as such, causes an enhancing effect on mineral deposit formation in the polymeric state. The most likely explanation for this amplifying effect of polyP is a local extrusion and sequestration of Ca2+-polyP from cells adjacent to the bone-forming sites of the osteoblasts, which appears to be very likely [39]. The alkaline phosphatase activity is spatially localized in mitochondria [72] on the apical region of the (secretory) cell membranes [73]. As schematically outlined in Fig. 11, the extracellularly localized alkaline phosphatase, induced by polyP, in a positive auto-circle increasingly degrades Ca2+-polyP to Ca2+ and PO 3-4 , perhaps allowing Ca-carbonated phosphate to form onto the CaCO3 bio-seeds. The exchange of ions is by far energetically favored in amorphous carbonated phosphate; such a state is likely to exist for this mineral. Interestingly enough, PO4 3− enzymatically released from polyP negatively controls CaCO3 formation by inhibiting the CA [34].

Schematic representation of the early stages of the enzymatic synthesis of the bio-seeds formed during bone mineralization. Two osteoblasts are depicted, one of them (upper) being involved in polyP metabolism. After enzymatic polyP formation and its compartmentalization into vesicles, the charges of the polymer are neutralized by cations, e.g., Ca2+. Those vesicles are extruded and polyP (Ca2+ salt) is discharged. Driven by the membrane-bound CA, HCO3 − and subsequently CaCO3 deposits are formed, which act as bio-seeds for calcium carbonate/phosphate and hydroxyapatite deposition. PolyP activates alkaline phosphatase. The released orthophosphate, in turn, inhibits the CA. The CA on the other side is activated by HCO3 − and by S. domuncula extract and quinolinic acid. PolyP polyphosphate, ATP adenosine triphosphate, ADP adenosine diphosphate, ALP alkaline phosphatase, CA carbonic anhydrase, NBC Na+-driven Cl−/HCO3 − exchanger, AE Na+-independent Cl−/HCO3 − exchanger
The data are schematically summarized in Fig. 11. For reasons of simplicity, the polyP (Ca2+ salt) storing cell is separated from the biomineralizing bone cell. PolyP is synthesized/degraded by a series of enzymes [74] and stored in vesicles as polyP (Ca2+ salt). The vesicles are sequestered into the extracellular space, where polyP induces the gene expression of alkaline phosphatase. This enzyme in turn accelerates the hydrolysis of polyP (Ca2+ salt), resulting in a spatial upregulation of the concentration of PO4 3− and Ca2+, the building blocks for Ca-phosphate formation. The adjacent bone mineralizing cell again spatially and locally synthesizes CaCO3 bio-seeds in close association of the cell membrane, either extracellularly or intracellularly under the control of CA. This enzyme catalyzes the rate-limiting step of HCO3 − formation. The exchange of HCO3 − between the extracellular and intracellular compartments is made possible by transporters and exchangers, e.g., NBC or AE. After formation of the CaCO3 bio-seeds, the activity of CA is downregulated by PO4 3−, as outlined before [38]. In the subsequent step the HCO3 − bio-seeds are partially transformed to Ca-carbonated phosphate, followed by deposition of Ca-phosphate. The activity of the two regulator enzymes, alkaline phosphatase and CA, is under feedback control, offering new targets for pharmacological intervention of bone diseases. While the alkaline phosphatase is activated by polyP, CA is inhibited by PO4 3−. Besides the physiological control of the enzyme activities, CA can be stimulated by secondary metabolites, produced by sponges, e.g., QA. Future studies will be performed to resolve the regulatory cross-talk between these two enzymes, CA and alkaline phosphatase, during the initial phase of bio-seed formation.
References
Xie B, Nancollas GH (2010) How to control the size and morphology of apatite nanocrystals in bone. Proc Natl Acad Sci USA 107:22369–22370
Beniash E (2011) Biominerals—hierarchical nanocomposites: the example of bone. Wiley Interdiscip Rev Nanomed Nanobiotechnol 3:47–69
Posner AS, Duyckaerts (1954) Infrared study of the carbonate in bone, teeth and francolite. Experientia 10:424–425
Posner AS (1969) Crystal chemistry of bone mineral. Physiol Rev 49:760–792
Biltz RM, Pellegrino ED (1977) The nature of bone carbonate. Clin Orthop 129:279–292
Poyart CF, Bursaux E, Fréminet A (1975) The bone CO2 compartment: evidence for a bicarbonate pool. Respir Physiol 25:89–99
Mann S, Parker SB, Ross MD, Skarnulis AJ, Williams RJ (1983) The ultrastructure of the calcium carbonate balance organs of the inner ear: an ultra-high resolution electron microscopy study. Proc R Soc Lond B 218:415–424
Pisam M, Jammet C, Laurent D (2002) First steps of otolith formation of the zebrafish: role of glycogen? Cell Tissue Res 310:163–168
Andrade LR, Lins U, Farina M, Kachar B, Thalmann R (2012) Immunogold TEM of otoconin 90 and otolin—relevance to mineralization of otoconia, and pathogenesis of benign positional vertigo. Hear Res 292:14–25
Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris RC Jr (1994) Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N Engl J Med 330:1776–1781
De Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM (2009) Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20:2075–2084
Bachra BN, Trautz OR, Simon SL (1963) Precipitation of calcium carbonates and phosphates. I. Spontaneous precipitation of calcium carbonates and phosphates under physiological conditions. Arch Biochem Biophys 103:124–138
Roos A, Boron WF (1981) Intracellular pH. Physiol Rev 61:296–434
Haddad GG, Boron WF (2000) Na+/HCO3 − cotransporters in rat brain: expression in glia, neurons, and choroid plexus. J Neurosci 20:6839–6848
Lindsey AE, Schneider K, Simmons DM, Baron R, Lee BS, Kopito RR (1990) Functional expression and subcellular localization of an anion exchanger from choroid plexus. Proc Natl Acad Sci USA 87:5278–5282
Casey JR, Sly WS, Shah GN, Alvarez BV (2009) Bicarbonate homeostasis in excitable tissues: role of AE3 Cl−/HCO3− exchanger and carbonic anhydrase XIV interaction. Am J Physiol Cell Physiol 297:C1091–C1102
Wilbur KM, Jodrey LH (1955) Studies on shell formation. V. The inhibition of shell formation by carbonic anhydrase inhibitors. Biol Bull 108:359–365
Berg JT, Ramanathan S, Gabrielli MG, Swenson ER (2004) Carbonic anhydrase in mammalian vascular smooth muscle. J Histochem Cytochem 52:1101–1106
Alvarez L, Fanjul M, Carter N, Hollande E (2001) Carbonic anhydrase II associated with plasma membrane in a human pancreatic duct cell line (CAPAN-1). J Histochem Cytochem 49:1045–1053
Purkerson JM, Schwartz GJ (2005) Expression of membrane-associated carbonic anhydrase isoforms IV, IX, XII, and XIV in the rabbit: induction of CA IV and IX during maturation. Am J Physiol Regul Integr Comp Physiol 288:R1256–R1263
Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, Stevens MM (2012) The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci USA 109:14170–14175
Omelon S, Georgiou J, Henneman ZJ, Wise LM, Sukhu B, Hunt T, Wynnyckyj C, Holmyard D, Bielecki R, Grynpas MD (2009) Control of vertebrate skeletal mineralization by polyphosphates. PLoS One 4:e5634
Posner AS, Betts F, Blumenthal NC (1978) Properties of nucleating systems. Metab Bone Dis Relat Res 1:179–183
Wang XH, Schröder HC, Wang K, Kaandorp JA, Müller WEG (2012) Genetic, biological and structural hierarchies during sponge spicule formation: from soft sol-gels to solid 3D silica composite structures. Soft Matter 8:9501–9518
Müller WEG, Schröder HC, Burghard Z, Pisignano D, Wang XH (2013) Silicateins—a novel paradigm in bioinorganic chemistry: enzymatic synthesis of inorganic polymeric silica. Chem Eur J 19:5790–5804
Peters A, Korte C, Hesse D, Zakharov N, Janek J (2007) Ionic conductivity and activation energy for oxygen ion transport in superlattices—the multilayer system CSZ (ZrO2 + CaO)/Al2O3. Solid State Ion 178:67–76
Zhu Z, Xue LM, Han T, Jiao L, Qin LP, Li YS, Zheng HC, Zhang QY (2010) Antiosteoporotic effects and proteomic characterization of the target and mechanism of an Er-Xian decoction on osteoblastic UMR-106 and osteoclasts induced from RAW264.7. Molecules 15:4695–4710
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE (1983) Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA 80:2752–2756
Shinohara C, Yamashita K, Matsuo T, Kitamura S, Kawano F (2007) Effects of carbonic anhydrase inhibitor acetazolamide (AZ) on osteoclasts and bone structure. J Hard Tissue Biol 16:115–123
Supuran CT, Scozzafava A (2000) Carbonic anhydrase inhibitors and their therapeutic potential. Expert Opin Ther Patents 10:575–600
Pastorekova S, Parkkila S, Pastorek J, Supuran CT (2004) Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 19:199–229
Supuran CT (2008) Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 7:168–181
Supuran CT (2008) Carbonic anhydrases—an overview. Curr Pharm Des 14:603–614
Müller WEG, Schröder HC, Schlossmacher U, Grebenjuk VA, Ushijima H, Wang XH (2013) Induction of carbonic anhydrase in SaOS-2 cells, exposed to bicarbonate and consequences for calcium phosphate crystal formation. Biomaterials 34:8671–8680
Müller WEG, Schröder HC, Schlossmacher U, Neufurth M, Geurtsen W, Korzhev M, Wang XH (2013) The enzyme carbonic anhydrase as an integral component of biogenic Ca-carbonate formation in sponge spicules. FEBS Open Bio 3:357–362
Müller WEG, Schlossmacher U, Schröder HC, Lieberwirth I, Glasser G, Korzhev M, Neufurth M, Wang XH (2013) Enzyme-accelerated and structure-guided crystallization of Ca-carbonate: role of the carbonic anhydrase in the homologous system. Acta Biomater 10:450–462
Müller WEG, Wiens M, Adell T, Gamulin V, Schröder HC, Müller IM (2004) Bauplan of Urmetazoa: basis for genetic complexity of Metazoa. Int Rev Cytol 235:53–92
Müller WEG, Wang XH, Diehl-Seifert B, Kropf K, Schloßmacher U, Lieberwirth I, Glasser G, Wiens M, Schröder HC (2011) Inorganic polymeric phosphate/polyphosphate as an inducer of alkaline phosphatase and a modulator of intracellular Ca2+ level in osteoblasts (SaOS-2 cells) in vitro. Acta Biomater 7:2661–2671
Mahamid J, Sharir A, Gur D, Zelzer E, Addadi L, Weiner S (2011) Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: a cryo-electron microscopy study. J Struct Biol 174:527–535
Sadan O, Shemesh N, Barzilay R, Dadon-Nahum M, Blumenfeld-Katzir T, Assaf Y, Yeshurun M, Djaldetti R, Cohen Y, Melamed E, Offen D (2012) Mesenchymal stem cells induced to secrete neurotrophic factors attenuate quinolinic acid toxicity: a potential therapy for Huntington’s disease. Exp Neurol 234:417–427
Gilis M, Grauby O, Willenz P, Dubois P, Heresanu V, Baronnet A (2013) Biomineralization in living hypercalcified demosponges: toward a shared mechanism? J Struct Biol 183:441–454
Fogh J, Fogh JM, Orfeo T (1977) One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59:221–226
Mikami T, Koyama T, Koyama T, Imakiire A, Yamamoto K, Furuhata M, Toyota H, Mizuguchi J (2006) C-jun N-terminal kinase activation is required for apoptotic cell death induced by TNF-related apoptosis-inducing ligand plus DNA-damaging agents in sarcoma cell lines. Anticancer Res 26:1153–1160
Schröder HC, Borejko A, Krasko A, Reiber A, Schwertner H, Müller WEG (2005) Mineralization of SaOS-2 cells on enzymatically (silicatein) modified bioactive osteoblast-stimulating surfaces. J Biomed Mater Res B Appl Biomater 75:387–392
Müller WEG, Boreiko A, Wang XH, Krasko A, Geurtsen W, Custódio MR, Winkler T, Lukić-Bilela L, Link T, Schröder HC (2007) Morphogenetic activity of silica and bio-silica on the expression of genes, controlling biomineralization using SaOS-2 cells. Calcif Tissue Int 81:382–393
Wiens M, Wang XH, Schlossmacher U, Lieberwirth I, Glasser G, Ushijima H, Schröder HC, Müller WEG (2010) Osteogenic potential of bio-silica on human osteoblast-like (SaOS-2) cells. Calcif Tissue Int 87:513–524
Wiens M, Wang XH, Schröder HC, Kolb U, Schlossmacher U, Ushijima H, Müller WEG (2010) The role of biosilica in the osteoprotegerin/RANKL ratio in human osteoblastlike cells. Biomaterials 31:7716–7725
Gregory CA, Gunn WG, Peister A, Prockop DJ (2004) An alizarin red–based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem 329:77–84
Magid E (1968) The dehydration kinetics of human erythrocytic carbonic anhydrases B and C. Biochim Biophys Acta 151:236–244
Carter ND, Chegwidden WR, Hewett-Emmett D, Jeffery S, Shiels A, Tashian RE (1984) Novel inhibition of carbonic anhydrase isozymes I, II and III by carbamoyl phosphate. FEBS Lett 165:197–200
Slowinski EJ, Wolsey WC, Masterton WL (2009) Chemical principles in the laboratory, 9th edn. Brooks/Cole, Belmont
Schröder HC, Sudek S, De Caro S, De Rosa S, Perović S, Steffen R, Müller IM, Müller WEG (2002) Synthesis of the neurotoxin quinolinic acid in apoptotic tissue from Suberites domuncula: cell biological, molecular biological and chemical analyses. Mar Biotechnol 4:546–558
Reinhard JF, Erickson JB, Flanagan EM (1994) Quinolinic acid in neurological disease: opportunities for novel drug discovery. Adv Pharmacol 30:85–127
Salge T, Terborg R (2009) EDS microanalysis with the silicon drift detector (CDD): innovative analysis options for mineralogical and material science application. Anadolu Univ J Sci Technol 10:45–55
Sachs L (1984) Angewandte Statistik. Springer, Berlin
Raz S, Testeniere O, Hecker A, Weiner S, Luquet G (2002) Stable amorphous calcium carbonate is the main component of the calcium storage structures of the crustacean Orchestia cavimana. Biol Bull 203:269–274
Mahamid J, Aichmayer B, Shimoni E, Ziblat R, Li C, Siegel S, Paris O, Fratzl P, Weiner S, Addadi L (2010) Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc Natl Acad Sci USA 107:6316–6321
Boskey AL, Posner AS (1973) Conversion of amorphous calcium phosphate to microcrystalline hydroxyapatite: a pH-dependent, solution-mediated, solid–solid conversion. J Phys Chem 77:2313–2317
Nudelman F, Pieterse K, George A, Bomans PH, Friedrich H, Brylka LJ, Hilbers PA, de With G, Sommerdijk NA (2010) The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat Mater 9:1004–1009
Dey A, Bomans PH, Müller FA, Will J, Frederik PM, de With G, Sommerdijk NA (2010) The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat Mater 9:1010–1014
Bentov S, Weil S, Glazer L, Sagi A, Berman A (2010) Stabilization of amorphous calcium carbonate by phosphate rich organic matrix proteins and by single phosphoamino acids. J Struct Biol 171:207–215
Querido W, Abraçado LG, Rossi AL, Campos APC, Fragoso CLR, Farina M (2011) Characterization of the bone-like apatite produced in a novel model for bone mineralization under treatment with strontium ranelate. Microsc Acta Suppl 20B:1–2
Rey C, Kim HM, Gerstenfeld L, Glimcher MJ (1996) Characterization of the apatite crystals of bone and their maturation in osteoblast cell culture: comparison with native bone crystals. Connect Tissue Res 35:343–349
Leyhausen G, Lorenz B, Zhu H, Geurtsen W, Bohnensack R, Müller WEG, Schröder HC (1998) Inorganic polyphosphate in human osteoblast-like cells. J Bone Miner Res 13:803–812
Hofmann LC, Straub S, Bischof K (2013) Elevated CO2 levels affect the activity of nitrate reductase and carbonic anhydrase in the calcifying rhodophyte Corallina officinalis. J Exp Bot 64:899–908
Sarma AS, Daum T, Müller WEG (1993) Secondary metabolites from marine sponges., Part I: Origin and chemistry of new metabolites, and synthetic studies. Part II: Biological properties of new metabolites and physiological activities of avarol and related compounds isolated from Dysidea sp.Akademie gemeinnütziger Wissenschaften zu Erfurt, Ullstein-Mosby Verlag, Berlin
Bhakuni DS, Rawat DS (2005) Bioactive marine natural products. Springer, New York
Lindskog S (1997) Structure and mechanism of carbonic anhydrase. Pharmacol Ther 74:1–20
Ilies M, Banciu MD, Ilies MA, Scozzafava A, Caproiu MT, Supuran CT (2002) Carbonic anhydrase activators: design of high affinity isozymes I, II and IV activators, incorporating tri-/tetrasubstituted-pyridinium-azole moieties. J Med Chem 45:504–510
An Z (2009) Bis(μ-2′-carboxylatobiphenyl-2-carboxylic acid-κ2O2:O2′)bis[(2,2′-bipyridine-κ2 N, N′)(2′-carboxylatobiphenyl-2-carboxylic acid-κO2′)zinc(II)]. Acta Crystallogr E E65:m1501
Sei Y, Fossom L, Goping G, Skolnick P, Basile AS (1998) Quinolinic acid protects rat cerebellar granule cells from glutamate-induced apoptosis. Neurosci Lett 241:180–184
Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gómez-García MR, Abramov AY (2010) Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem 285:9420–9428
Wang H, Zhang P, Liu L, Zou L (2013) Hierarchical organization and regulation of the hematopoietic stem cell osteoblastic niche. Crit Rev Oncol Hematol 85:1–8
Rao NN, Gómez-García MR, Kornberg A (2009) Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78:605–647
Ilies M, Scozzafava A, Supuran CT (2004) Carbonic anhydrase activators. In: Supuran CT, Scozzafava A, Conway J (eds) Carbonic anhydrase—its inhibitors and activators. CRC Press, Boca Raton, pp 317–352
Acknowledgments
W. E. G. M. is a holder of an ERC Advanced Investigator Grant (268476 BIOSILICA). This work was supported by grants from the Deutsche Forschungsgemeinschaft (Schr 277/10-3), the European Commission (“Bio-Scaffolds-Customized Rapid Prototyping of Bioactive Scaffolds,” 604036; Industry-Academia Partnerships and Pathways “CoreShell,” 286059; “MarBioTec*EU-CN*,” 268476; and “BlueGenics,” 311848), and the International Human Frontier Science Program.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, X., Schröder, H.C., Schlossmacher, U. et al. Modulation of the Initial Mineralization Process of SaOS-2 Cells by Carbonic Anhydrase Activators and Polyphosphate. Calcif Tissue Int 94, 495–509 (2014). https://doi.org/10.1007/s00223-013-9833-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-013-9833-4