
Abstract
Uncertainties exist regarding whether FGF-23 production is influenced by PTH and its involvement in bone formation. We evaluated FGF-23 response and its relation to changes in biomarkers of bone formation following intermittent PTH treatment. Twenty-seven women with a mean [SD] age of 75.8 [5.4] years with postmenopausal osteoporosis were treated with PTH(1–34) for 18 months. Bone mineral density (BMD) was measured at 6 and 18 months at the lumbar spine (LS) and total hip (TH). Blood samples were obtained at baseline, 1–3, 6–9, and 12–18 months. Serum calcium, phosphate, PTH, 25(OH)vitamin D, 1,25(OH)2vitamin D, markers of bone turnover, FGF-23, and sclerostin were measured. BMD increased at both the LS (11.6%, P < 0.001) and TH (2.5%, P < 0.01). The bone formation marker P1NP increased early (baseline mean [SD] 39.9 [24.4] μg/l, 1–3 months 88 [37.9] μg/l; P < 0.001) and remained higher than baseline throughout 18 months. FGF-23 also increased, with a peak response at 6–9 months (increase 65%, P = 0.002). Serum phosphate remained stable. A significant increase in 1.25(OH)2vitamin D (P = 0.02) was seen at 1–3 months only. A small but significant reduction in sclerostin was seen at 6–9 (P = 0.02) and 12–18 months (P = 0.06). There was a positive correlation between changes in P1NP and FGF-23 (6–9 months r = 0.78, P < 0.001). FGF-23 is increased by intermittent PTH(1–34). This is related to early changes in P1NP, suggesting that the skeletal effects of PTH may involve FGF-23. Further studies are required to elucidate this.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fibroblast growth factor-23 (FGF-23) is a phosphaturic factor, implicated in several phosphate disorders [1, 2]. It is produced by osteoblasts and osteocytes [3]. Both phosphate and 1,25(OH)2 vitamin D have been shown to regulate its production [4–7]. FGF-23 downregulates the activity of the sodium/phosphate cotransporters NaPi2a and NaPi2c, which promotes urinary phosphate excretion [8]. 1,25(OH)2 Vitamin D upregulates FGF-23, which then acts in a regulatory loop to reduce the synthesis of 1,25(OH)2 vitamin D [9]. The regulation of FGF-23 by 1,25(OH)2 vitamin D may be phosphate-independent [6].
It is unclear whether PTH has a direct role in the regulation of FGF-23 production [10, 11]. In pathological states including primary and secondary hyperparathyroidism, such as in chronic kidney disease (CKD) [11–13], PTH concentrations have been associated with increases in FGF-23 in some, but not all, studies [13, 14]. Both factors are thought to act synergistically to promote urinary phosphate loss [6]. The marked elevations in circulating FGF-23 observed in CKD are consistent with increasing FGF-23 expression in bone [15] and are associated with PTH concentrations, suggesting that the expression of FGF-23 in bone, at least in CKD, may be regulated partially by a PTH-dependent pathway [16]. Large population-based studies have shown a positive correlation between serum FGF-23 and PTH in subjects without CKD [17]. PTH injection in mice has been shown to increase FGF-23 [18]. Furthermore, in a study of healthy men, PTH infusion at pharmacological doses increased serum FGF-23 concentrations and endogenous 1,25(OH)2 vitamin D [19]. However, the authors were unable to determine whether the increases in FGF-23 were due to a direct effect of PTH, independently of 1,25(OH)2vitamin D.
Uncertainties also remain about the effect of FGF-23 on bone formation and remodeling. FGF-23 expression in bone is closely related to bone formation in vivo and in vitro [20, 21]. Overexpression of FGF-23 during osteoblast development had no effect on osteoprogenitor proliferation but markedly suppressed differentiation and matrix mineralization [21]. Severe mineralization defects have been observed in vivo in a mouse model deficient in both FGF-23 and NaPi2a genes, suggesting that FGF-23 is required for mineralization, independently of its systemic effects on phosphate homeostasis [22]. In children and young adults with end-stage renal disease (ESRD), increased circulating FGF-23 and bone expression have been related to improved bone mineralization indices [23]. Although these data suggest a possible role for FGF-23 in the local regulation of skeletal mineralization in renal bone disease, further studies on the effect of FGF-23 in skeletal remodeling are needed.
Intermittent PTH treatment increases osteoblast numbers [24] and affects osteoblast differentiation [25]. Recent data also indicate that the bone formation activity of PTH involves the osteocytes [26]. PTH administration in mice has been shown to downregulate sclerostin expression [27], which is an antagonist of the Wnt signaling pathway and an inhibitor of osteoblast formation and differentiation. Activation of PTH-R1 signaling in osteocytes inhibits sclerostin and increases bone formation [28]. It therefore appears plausible that the production of FGF-23, another factor produced by osteocytes, may be influenced by intermittent PTH.
In order to address the uncertainties regarding the role of (1) PTH in FGF-23 production/secretion and (2) FGF-23 in bone formation, we evaluated the regulation of FGF-23 and its relation to changes in markers of bone formation following intermittent PTH treatment in postmenopausal osteoporosis.
Materials and Methods
Subjects
Thirty-five women with established postmenopausal osteoporosis who met the criteria for treatment eligibility with intermittent PTH were invited to take part in the study. Twenty-seven women with a mean [SD] age of 75.8 [5.4] years agreed and were enrolled in the study. All were eligible for treatment with the PTH analogue (1–34) according to the guidance set out by the National Institute for Clinical Excellence (NICE), which included previous fragility fracture, insufficient response, intolerance to oral bisphosphonate, and low BMD (T score <–4.0 or <–3.0 plus 2 or more fractures) and at least one independent risk factor. All patients had osteoporosis and previous fragility fractures. All had received previous treatment with an oral bisphosphonate with a duration of (mean [SD]) of 4 [2] years. Fifteen patients were receiving alendronate 70 mg weekly, and the rest (n = 12) were on risedronate 35 mg weekly. Oral bisphosphonates were stopped prior to teriparatide treatment as detailed below. Twelve patients (43%) had become intolerant of oral bisphosphonates and developed clinically significant upper gastrointestinal symptoms, and 15 (57%) had had an unsatisfactory response to oral bisphosphonates as defined by a fall in BMD or a new fragility fracture despite treatment with bisphosphonate. Sixteen patients had previously sustained at least one vertebral fracture, and 24 patients had a previous history of peripheral fractures. The patient demographics and baseline biochemical parameters are summarized in Table 1. The study was approved by the Local Research Ethics Committee of Guy’s and St. Thomas’ Hospital NHS Trust, and all patients gave informed consent. This was a prospective study of treatment with intermittent PTH in the clinical setting of a routine metabolic bone clinic.
At the time of study entry, all patients stopped oral bisphosphonates and were started on teriparatide (20 μg/day subcutaneously) for 18 months. There was no washout period. They were asked to continue the calcium (1,000 mg/day) and vitamin D3 (800 IU/day) supplements in addition to teriparatide. Random nonfasting blood and spot urine samples were collected at baseline. Routine biochemical parameters were analyzed immediately, while additional serum and plasma samples were frozen and stored at –70°C for subsequent analysis of FGF-23, 1,25(OH)2 vitamin D, and the bone turnover marker total procollagen type 1 amino-terminal propeptide (P1NP). Blood samples were obtained at least 6 hours after subcutaneous injection of teriparatide. Samples were taken at subsequent clinic visits and grouped as 1–3, 6–9, and 12–18 months. Data were grouped in this manner to account for variability within 1 month in the dates of outpatient appointments. Bone mineral density (BMD) was assessed at the total hip (TH) and lumbar spine (LS). This was measured at baseline and at 6 and 18 months following teriparatide treatment.
FGF-23 was also measured in 20 postmenopausal women aged 66.1 [6.8] years who were taking part in a study looking at the effects of discontinuation of oral bisphosphonates for 12 months, after long-term use, on bone metabolism. The subjects had been on bisphosphonates for 5.0 [1.8] years. All subjects gave informed consent. All were asked to continue with the calcium and vitamin D supplements as above. Half (n = 10) of the subjects had been on alendronate, and the rest were receiving risedronate. Their serum 25(OH) vitamin D and PTH concentrations at the time of bisphosphonate withdrawal were 71.6 [23] nmol/l and 31 [12] ng/l, respectively.
Laboratory Measurements
Routine biochemical tests including serum calcium, albumin, phosphate, and creatinine were measured by standard laboratory methods on Roche Modular analyzers (Roche Diagnostics, Burgess Hill, UK). Estimated glomerular filtration rate (eGFR) was derived using the Modification of Diet in Renal Disease (MDRD) formula [29]. Serum intact PTH was measured by electrochemiluminescence immunoassay on the Roche Elecsys 2010 analyzer (Roche Diagnostics, Indianapolis, IN). 25-Hydroxyvitamin D (25[OH] vitamin D) was measured by radioimmunoassay (RIA) (Diasorin, Stillwater, MN). Interassay coefficient of variation (CV) was <10% at serum 25(OH) vitamin D concentrations of 40 and 131.0 nmol/l. 1,25(OH)2 Vitamin D was measured by RIA (Immunodiagnostic Systems, Boldon, UK) as previously described [30].
The bone formation marker P1NP was measured on the Elecsys 2010 automated platform by an electrochemiluminescence immunoassay. The intra-assay CV was <2.5% at concentrations of 66, 358, and 707 μg/l. Bone-specific alkaline phosphatase (BALP) was measured by an immunoenzymetric assay (Immunodiagnostic Systems). Assay CV was 1.9 and 9% at BALP concentrations of 10.6 and 42.7 μg/l, respectively. Intact osteocalcin was measured by the Metra Osteocalcin immunoassay (Quidel, San Diego, CA, USA). Between-batch CVs were <10% at osteocalcin concentrations of 6.2, 7.4, and 16.6 ng/ml. Urine CTX was measured on a single random urine sample by ELISA (Urine Crosslaps; Nordic Bioscience Diagnostics, Herlev, Denmark). The premenopausal reference range was 45–1,035 μg/mmol creatinine. Interassay CV was <10%.
FGF-23 was measured by a two-site enzyme-linked immunosorbent assay (ELISA) (Immunotopics, San Clemente, CA), which detects both the intact FGF-23 and the C-terminal fragments. A strong linear correlation has been observed between the C-terminal and the intact assays. Intra-assay CVs were 8.9 and 8.6% and interassay CVs were 7.7 and 8.3% at serum concentrations of 37 and 370 RU/l, respectively. The detection limit of the assay was 3.0 RU/l. Sclerostin was measured by ELISA (prelaunch version; Biomedica Gruppe, Vienna, Austria). The assay range was 0–80 pmol/l. Inter- and intra-assay CVs were 4% at a serum concentration of 17.2 pmol/l. The detection limit of the assay was 2.0 pmol/l. To minimize variability, samples obtained from the same patient at the different time points were analyzed for FGF-23 and sclerostin in the same batch.
BMD
BMD at the LS and TH was measured by dual-energy X-ray absorptiometry (Discovery-A QDR Series; Hologic, Waltham, MA). The CV for the BMD measurements was 0.35%.
Statistical Analyses
Statistical analysis was performed using SPSS 17.0 for Windows (Leadtools; LEAD Technologies Inc, Charlotte, NC, USA). All values are represented as mean ± SD unless stated otherwise. Values for FGF-23, 1,25(OH)2 vitamin D, and sclerostin are shown as mean [SEM]. The paired t-test was used to compare the biochemical data at the different time points to baseline values. The unpaired t-test was used to compare data between two groups. Nonparametric data were log-transformed. Univariate analysis (Pearson’s correlation coefficient) was performed to look for correlation between FGF-23 and P1NP. P < 0.05 was considered significant.
Results
BMD
BMD at the LS increased significantly at 6 months (mean [SEM] 6.5% [0.8%]). In contrast, a small early decline in BMD at the TH was seen at 6 months, although this was not significant (–0.5% [0.7%]). LS BMD increased further at 18 months, with increases of 11.6% [0.9%] compared to baseline. A significant increase in TH BMD was seen at 18 months (2.5% [0.65%]), although the increase was less than that observed at the LS. There was a significant difference in percent change in BMD at the LS only at 6 months between the group previously treated with alendronate and the risedronate-treated group (alendronate 4.72 [0.8] vs. risedronate 8.54 [1.4], P = 0.03). No significant difference in the BMD response was seen between the two groups after 18 months’ treatment with teriparatide.
Changes in Serum Calcium, Phosphate, PTH, and 25(OH) Vitamin D
Serum total calcium increased significantly from baseline at 1–3 (P = 0.016) and 6–9 (P < 0.001) months, although the mean serum concentrations remained within the normal range. This is summarized in Table 2. Mild transient hypercalcemia (albumin-adjusted calcium >2.55 mmol/l) was observed in three patients at 1–3 months (mean [SEM] 2.64 [0.03] mmol/l) and in six patients at 6–9 months (2.65 [0.01] mmol/l). The calcium supplements were reduced for 3 months until their next clinic visit. Only one patient was noted to still have mild hypercalcemia at 12 months. PTH decreased significantly from baseline at 1–3 and 6–9 months (P < 0.01). No significant change in serum phosphate and eGFR was seen at any time points. GFR was 79.4 [3.7] ml/min at baseline and 75.7 [4] ml/min at the end of the study. Serum 25(OH) vitamin D decreased significantly at all time points compared to baseline (Table 2).
Changes in the Biochemical Markers of Bone Turnover
P1NP increased significantly as early as 1–3 months by 121% with a peak response seen at 6–9 months (274%) and remained higher than at baseline at 12–18 months (Table 3). There was no significant difference in P1NP at 6–9 months and at 12–18 months (mean [SEM] 6–9 months 149 [17], 12–18 months 121.4 [11] μg/l). At 1–3 months, all patients had an increase in P1NP of >10 μg/l. Early increases in the other markers of bone formation, osteocalcin and BALP, were seen in the whole study population (Table 3). The marker of bone resorption urine CTX showed a delayed increase compared to the bone formation markers. No significant difference in the increase in P1NP was observed at any time points between the patients previously on alendronate compared to those on risedronate.
Changes in Serum 1,25(OH)2 Vitamin D, FGF-23, and Sclerostin
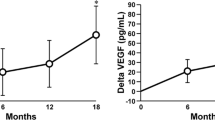
Serum 1,25(OH)2 vitamin D increased early at 1–3 months (baseline 114.6 [10.7], 1–3 months 132.4 [11] pmol/l, P = 0.02) and stayed elevated at 6–9 months (140 [17.8]), although this was not significant (P = 0.1) (Fig. 1). FGF-23 increased significantly from baseline at 1–3 months (33%, P = 0.007), 6–9 months (63%, P = 0.002), and 12–18 months (58%, P = 0.03) and paralleled the increase in P1NP (Fig. 2). No significant difference in FGF-23 response was seen between the previously alendronate-treated group compared to the risedronate-treated group. Four patients had an elevated baseline FGF-23 concentration (628.4 [94.4] RU/l). FGF-23 concentrations increased in these four patients following teriparatide, with a mean percent rise of 45% seen at 6–9 months. The baseline serum phosphate, 25(OH) vitamin D, 1,25 (OH)2 vitamin D, PTH, and eGFR of the four patients did not differ significantly from the rest of the study group. However, we observed a lower hemoglobin concentration (mean [SD] 11.6 [1.6] g/dl) and low serum iron concentration (5.0 [1.8] μmol/l, normal range 5.4–28.8 μmol/l) in these four patients. In contrast to FGF-23, a small reduction in sclerostin was observed at 6–9 months (P = 0.02) and at 12–18 months, although the results just failed to reach significance (P = 0.06) (Fig. 3).

Changes in serum FGF-23 (RU/l) and 1,25(OH)2 vitamin D (pmol/l) following treatment with teriparatide. Values represent the mean [SEM]. A significant increase in 1,25(OH)2D was seen at 1–3 months. FGF-23 increased significantly at all time points. * P < 0.05, # P < 0.01 compared to baseline

Changes in FGF-23 (RU/l) and serum P1NP (μg/l) following treatment with teriparatide P1NP increased significantly at all time points, with a peak at 6–9 months. There was a parallel increase in FGF-23. * P < 0.05, # P < 0.01, ## P < 0.0001 compared to baseline

Changes in serum sclerostin and FGF-23 following treatment with teriparatide. A significant reduction in serum sclerostin was seen at 6–9 months (P = 0.02). * P < 0.05, # P < 0.01 compared to baseline
Circulating FGF-23 concentrations did not change significantly from baseline in the group who discontinued bisphosphonate but did not go on teriparatide (control group FGF-23 [RU/l] mean [SEM]: baseline 109.6 [27.5], 6 months 86.7 [23], 12 months 140 [28.3]).
Relationship Between Changes in FGF-23 and P1NP
Significant correlations were found between changes in FGF-23 and P1NP during treatment with teriparatide at 1–3 and 6–9 months (1–3 months r = 0.39, P < 0.05; 6–9 months r = 0.78, P < 0.001). A significant correlation was also seen between changes in FGF-23 and ALP at 6–9 months only (r = 0.43, P < 0.05). No significant correlation was observed between change in FGF-23 or osteocalcin and 1,25(OH)2 vitamin D at any time point.
Discussion
Data from this small prospective study are in keeping with the findings of larger randomized controlled trials [24]. We observed significant increases in P1NP and BMD at the LS and TH, with the increases at the TH seen later than those at the LS. We found an earlier significant increase in the bone formation markers compared to the resorption marker, in accordance with the “anabolic window” hypothesis [31]. A larger increase in LS BMD following PTH(1–34) for 6 months was seen in those patients who had had prior treatment with risedronate compared to those on prior alendronate. The BMD response, however, was similar in both groups after 18 months. Previous studies have indeed shown that the response to teriparatide may differ as a function of the previous type of bisphosphonate used [32, 33].
The patients were changed to anabolic treatment immediately after bisphosphonates had been ceased. This did not affect the responsiveness as we observed changes in BMD and bone turnover of the same order of magnitude as previously reported in treatment-naive patients [24]. Indeed, recent data imply that bisphosphonate washout does not appear to influence the treatment effect of teriparatide [34]. Some patients developed mild transient hypercalcemia, as previously documented [24]. Endogenous PTH decreased significantly throughout the 18 months compared to pretreatment serum concentrations. Serum phosphate did not change significantly following PTH(1–34) and remained in the normal range. This is consistent with previously reported data following PTH infusion [19]. Serum 25(OH) vitamin D decreased significantly at 3 months and remained lower than baseline concentration. A contributing factor may have been reduction in the intake of vitamin D as the patients who developed mild hypercalcemia were asked to reduce their calcium/vitamin D supplements. However, only one study participant reduced her calcium and vitamin D supplement throughout the study period. A possible explanation is accelerated catabolism of 25(OH) vitamin D by increased 1,25(OH)2 vitamin D as observed in patients with primary hyperparathyroidism [35].
Serum 1,25(OH)2 vitamin D increased early and paralleled the increase in FGF-23 at 1–3 months, suggesting that FGF-23 production may have been upregulated by 1,25(OH)2 vitamin D, at least at the early time point. Indeed, 1,25(OH)2 vitamin D is a powerful regulator of FGF-23 and acute increases in serum FGF-23 have been observed following calcitriol [6]. However, the elevations in circulating FGF-23 concentrations following PTH persisted throughout the 18 months. We did not see any difference in FGF-23 concentrations at 6–9 months between the group previously treated with alendronate compared to the risedronate group, although a significant difference in percent change in BMD at the LS was observed between the two groups. This may be due partly to the small number of subjects in each group and the variability in FGF-23 response.
Although FGF-23 increased, we did not observe a reduction in serum phosphate. This may be due, in part, to the observed reduction in endogenous PTH production as it is now thought that both PTH and FGF-23 are needed for the maximal phosphaturic effect of FGF-23. However, although PTH declined, the concentrations were higher than those seen in patients with hypoparathyroidism [6]; therefore, this cannot entirely explain our findings. In a recent study of healthy men, PTH infusion led to elevations in FGF-23 but, as in our study, no change in serum phosphate was observed [19]. One explanation is that the random measurement of serum phosphate is not sensitive enough to detect the transient changes in phosphate balance following intermittent PTH. Another explanation for the findings may be related to increases in 1,25(OH)2 vitamin D, which helped to maintain normal serum phosphate. Circulating FGF-23 concentrations, as measured by the C-terminal FGF-23 immunoassay, are elevated in patients with iron-deficiency anemia [36]. All four patients who had an elevated baseline FGF-23 in our study were noted to have low serum iron. These patients did not have evidence of hypophosphatemia. The increase in FGF-23 in these patients may be related to the accumulation of biologically inactive fragments in iron deficiency.
The issue of whether PTH modulates FGF-23 remains largely unresolved [37]. It may well be that at physiological concentrations PTH does not regulate FGF-23. However, when used intermittently, the anabolic effects of PTH on osteoblasts may lead to increased production of FGF-23. This may represent a different aspect of the effect of PTH on osteoblasts and/or osteocytes. In support of this concept is the observation that FGF-23 is elevated in patients with fibrous dysplasia [38], a disorder which mimics activation of the PTH pathway, due to increased production by osteogenic cells. Further evidence from recent studies demonstrates that constitutive activation of PTHR1 in osteocytes leads to upregulation of FGF-23 [39]. Plasma levels of FGF-23 have also been found to be high in a patient with Jansen metaphyseal dysplasia, which is due to constitutively active PTHR1 [40]. It is unlikely that the changes in FGF-23 are related to changes in bone turnover or phosphate metabolism following bisphosphonate withdrawal in the study participants as there was no change in FGF-23 in the subjects who discontinued oral bisphosphonate but were not treated with teriparatide. There have been reports, however, of a dose-dependent effect of bisphosphonate, disodium etidronate [41, 42], on the renal handling of phosphate by mechanisms which may be independent of the hormonal regulation of phosphate metabolism. It is possible that the newer nitrogen-containing bisphosphonates do not have the same effect on phosphate metabolism, particularly at the relatively small doses used in postmenopausal osteoporosis. Moreover, in a recent study [43], baseline circulating FGF-23 and increases in FGF-23 following PTH(1–34) infusion for 46 h did not differ between patients on dialysis with adynamic/normal bone histology and those with high turnover bone histology, suggesting that previous reduction of bone turnover with bisphosphonate is unlikely to affect the FGF-23 response to PTH.
Another area of uncertainty is whether the production of FGF-23 has a local effect on bone formation and mineralization. Several in vitro and in vivo studies have shown that FGF-23 may have an effect on bone formation/mineralization, independently of its effect on phosphate homeostasis [20, 22]. We observed a significant correlation between changes in FGF-23 and the bone formation marker P1NP. Previous studies have shown that continuous PTH infusion increases FGF-23 [19, 43]. Our data suggest that FGF-23 is also upregulated following intermittent PTH for 18 months. This stimulatory effect may be related to increases in osteoblastic activity. In support of this hypothesis, a previous report demonstrated a significant correlation between mineralization indices and high levels of FGF-23 in children with ESRD [23]. Interestingly, there is recent evidence to indicate that FGF-23 is regulated by osteoblastic bone formation and bone turnover rate [44]. In contrast to our study, the authors found that exogenous PTH administration reduced circulating concentrations of FGF-23. One explanation for the differences in their findings may be related to the timing of sampling. The reduction in FGF-23 was seen at one early time point only (day 20), and indeed, the reduction in FGF-23 was no longer significant at 3 months in their murine model. It is also possible that the reduction in FGF-23 in their animal model may be linked to suppression by 1,25(OH)2 vitamin D in an endocrine feedback loop between FGF-23 and 1,25(OH)2 vitamin D. Nevertheless, the data point to changes in FGF-23 during changes in bone remodeling and the possible involvement of this factor in mediating the bone anabolic effect of PTH. It is now becoming established that intermittent PTH suppresses the osteocytic expression of the bone formation antagonist sclerostin [45]. In our study, the increase in FGF-23 was paralleled by a decrease in circulating sclerostin. It is conceivable, therefore, that PTH may exert its cellular anabolic effects, at least partly, by stimulating the production of FGF-23. However, the effect of PTH on FGF-23 production should be viewed in a larger context with the possible involvement of a complex feedback loop between osteocytes, PTH, phosphate homeostasis, and possibly bone turnover. Further studies are required to investigate this possibility.
The findings of our study should be interpreted within their clinical context. The study was not a randomized controlled clinical trial and, thus, has its limitations compared to the controlled setting of animal models, described above [43]. It is also acknowledged that the association between changes in FGF-23 and P1NP does not prove causality.
In summary, our results show that in humans circulating concentrations of FGF-23 are increased by PTH, when given intermittently. A significant correlation was seen between FGF-23 and the bone formation marker P1NP, which raises the interesting possibility that FGF-23 may play a role in mediating the skeletal anabolic effects of PTH. However, further studies are required to investigate this novel potential physiological mechanism.
References
Consortium ADHR (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF-23. Nat Genet 26:345–348
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Tamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphataemia. N Engl J Med 348:1656–1663
Juppner H (2007) Novel regulators of phosphate homeostasis and bone metabolism. Ther Apher Dial 11(Suppl 1):S3–S22
Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N (2005) Circulating FGF-23 is regulated by 1alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 280:2543–2549
Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto KI (2005) Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab 288:E1101–E1109
Collins MT, Linsaay JR, Jain A, Kelly MH, Cutler CM, Weinstein LS, Liu J, Fedarko NS, Winer KK (2005) Fibroblast growth factor-23 is regulated by 1alpha, 25-dihydroxyvitamin D. J Bone Miner Res 20:1944–1950
Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, JSl Finkelstein (2006) Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21:1187–1196
Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M (2009) Fgf-23 decreases renal Na Pi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 297:F282–F291
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD (2006) Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 17:1305–1315
Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y (2007) Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol 18:2683–2688
Tebben PJ, Singh RJ, Clarke BL, Kumar R (2004) Fibroblast growth factor 23, parathyroid hormone, and 1alpha, 25-dIhydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc 79:1508–1513
Sato T, Tominaga Y, Ueki T, Goto N, Matsuoka S, Katayama A, Haba T, Uchida K, Nakanishi S, Kazama JJ, Gejyo F, Yamashita T, Fukagawa M (2004) Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis 44:481–487
Nakanishi S, Kazama JJ, Nii-Kono T, Omori K, Yamashita T, Fukumoto S, Gejyo F, Shigematsu T, Fukagawa M (2005) Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int 67:1171–1178
Urena Torres P, Friedlander G, de Vernejoul MC, Silve C, Prie D (2008) Bone mass does not correlate with the serum fibroblast growth factor 23 in hemodialysis patients. Kidney Int 73:102–107
Pereira RC, Juppner H, Azucena-Serrano CE, Yadin O, Salusky IB, Wesseling-Perry K (2009) Patterns of FGF-23, DMP1 and MEPE expression in patients with chronic kidney disease. Bone. doi:10.1016/j.bone.2009.08.008
Daji F, Shiizaki K, Shimada S, Okada T, Kunimoto K, Sakaguchi T, Hatamura I, Shigematsu T (2009) Regulation of fibroblast growth factor 23 production in bone in uremic rats. Nephron 111:61–68
Marsell R, Grundberg E, Krajisnik T, Mallmin H, Karlsson M, Mellstrom D, Orwoll E, Ohlsson C, Jonsson KB, Ljunggren O, Larsson TE (2008) Fibroblast growth factor-23 is associated with parathyroid hormone and renal function in a population-based cohort of elderly men. Eur J Endocrinol 158:125–129
Hasegawa H, Shimada T, Yamazaki Y (2004) Parathyroid-dependent and -independent mechanism to elevate serum concentration of FGF-23. J. Am Soc Nephrol 15:267A
Burnett-Bowie SM, Henao MP, Dere ME, Alaee H, Leder BZ (2009) Effects of hPTH(1–34) infusion on circulating serum phosphate, 1, 25-dihydroxyvitamin D and FGF23 levels in healthy men. J Bone Miner Res 24:1681–1685
Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N (2007) Mineralized tissue cells are a principal source of FGF23. Bone 40:1565–1573
Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N (2008) Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 23:939–948
Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schuler C, Erben RG, Lanske B (2008) Genetic evidence of serum phosphate-independent functions of fgf-23 on bone. PLOS Genet 4:e10000154
Wesseling-Perry K, Pereira RC, Wang H, Elashoff RM, Sahney S, Gales B, Jüppner H, Salusky IB (2009) Relationship between plasma FGF-23 concentration and bone mineralization in children with renal failure on peritoneal dialysis. J Clin Endocrinol Metab 94:511–517
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in post-menopausal women with osteoporosis. N Engl J Med 344:1434–1441
Jilka RL (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40:1434–1446
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37:148–158
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–4583
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3:e2942
Archibald G, Bartlett W, Brown A, Christie B, Elliott A, Griffith K, Pound S, Rappaport I, Robertson D, Semple Y, Slane P, Whitworth C, Williams B (2007) UK consensus conference on early chronic kidney disease–6 and 7. Nephrol Dial Transplant 22:2455–2457
Joseph F, Ahmad AM, Ul-Haq M, Durham BH, Whittingham P, Fraser WD, Vora JP (2008) Effects of growth hormone administration on bone mineral metabolism, PTH sensitivity and PTH secretory rhythm in postmenopausal women with established osteoporosis. J Bone Miner Res 23:721–729
Chen P, Miller PD, Delmas PD, Misurki DA, Krege JH (2006) Change in lumbar spine BMD and vertebral fracture risk reduction in teriparatide-treated postmenopausal women with osteoporosis. J Bone Miner Res 21:1785–1790
Miller PD, Delmas PD, Lindsay R, Watts NB, Luckey M, Adachi J, Saag K, Greenspan SL, Seeman E, Boonen S, Meeves S, Lang TF, Bilezikian JP (2008) Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 93:3785–3793
Russell RGG, Watts NB, Ebetino FH, Rogers MJ (2008) Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 19:733–759
Keel C, Kraenzlin ME, Kraenzlin CA, Muller B, Meier C (2010) Impact of bisphosphonate wash-out prior to teriparatide therapy in clinical practice. J Bone Miner Metab 28:68–76
Eastell R, Arnold A, Brandi ML, Brown EM, D’Amour P, Hanley DA, Sudhaker Rao D, Rubin MR, Goltzman D, Silverberg SJ, Marx SJ, Peacock M, Mosekilde L, Bouillon R, Lewiecki EM (2009) Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 94:340–350
Durham BH, Joseph F, Bailey LM, Fraser WD (2007) The association of circulating ferritin with serum concentrations of fibroblast growth factor-23 measured by 3 commercial assays. Ann Clin Biochem 44:463–466
Bergwitz C, Juppner H (2010) Regulation of phosphate homeostasis by PTH, vitamin D and FGF-23. Annu Rev Med 61:91–104
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey O (2003) FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112:683–692
Rhee Y, Farrow E, Lee R, Bivi N, Lazcano V, Plotkin L, White K, Bellido T (2009) FGF23 expression is upregulated by PTH receptor activation in osteocytes in vitro and in vivo: a parathyroid-bone link influencing the endocrine function of osteocytes. J Bone Miner Res 24 (suppl 1). http://www.asbmr.org/ Meetings/Annual meeting/A09001860 (accessed March 6, 2010)
Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, Mc Cormick KL (2008) Hypophosphatemia with elevations in FGF-23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab 94:17–20
Walton RJ, Russell RG, Smith R (1975) Changes in the renal and extrarenal handling of phosphate induced by disodium etidronate (EHDP) in man. Clin Sci Mol Med 49:45–56
Bonjour JP, Troehler U, Preston C, Fleisch H (1978) Parathyroid hormone and renal handling of Pi: effect of dietary Pi and diphosphonates. Am J Physiol Renal Physiol 234:F497–F505
Wesseling-Perry K, Harkins GC, Wang H, Elashoff R, Gales B, Horwitz MJ, Stewart AF, Juppner H, Salusky IB (2010) The calcemic response to continuous parathyroid hormone (PTH) (1–34) infusion in end-stage kidney disease varies according to bone turnover: a potential role for PTH (7–84). J Clin Endocrinol Metab 95:2772–2780
Samadfam R, Richard C, Nguyen-Yamamoto L, Bolivar I, Goltzman D (2009) Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology 150:4835–4845
Bellido T (2006) Downregulation of SOST/Sclerostin by PTH: a novel mechanism of hormonal control of bone formation mediated by osteocytes. J Musculoskelet Neuronal Interact 6:358–359
Author information
Authors and Affiliations
Corresponding author
Additional information
W D Fraser has received lecture fees and educational grant funding from Eli Lilly and Company. All other authors have stated that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Sridharan, M., Cheung, J., Moore, A.E. et al. Circulating Fibroblast Growth Factor-23 Increases Following Intermittent Parathyroid Hormone (1–34) in Postmenopausal Osteoporosis: Association with Biomarker of Bone Formation. Calcif Tissue Int 87, 398–405 (2010). https://doi.org/10.1007/s00223-010-9414-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-010-9414-8