
Abstract
Protein glycosylation is a highly heterogeneous post-translational modification that has been demonstrated to exhibit significant variations in various diseases. Due to the differential patterns observed in disease and healthy populations, the glycosylated proteins hold promise as early indicators for multiple diseases. With the continuous development of liquid chromatography-mass spectrometry (LC–MS) technology and spectrum analysis software, the sensitivity for the decipher of the tandem mass spectra of the glycopeptides carrying intact glycans, i.e., intact glycopeptides, enzymatic hydrolyzed from glycoproteins has been significantly improved. From quantified intact glycopeptides, the difference of protein glycosylation at multiple levels, e.g., glycoprotein, glycan, glycosite, and site-specific glycans, could be obtained for different samples. However, the manual analysis of the intact glycopeptide quantitative data at multiple levels is tedious and time consuming. In this study, we have developed a software tool named “GP-Marker” to facilitate large-scale data mining of spectra dataset of intact N-glycopeptide at multiple levels. This software provides a user-friendly and interactive interface, offering operational tools for machine learning to researchers without programming backgrounds. It includes a range of visualization plots displaying differential glycosylation and provides the ability to extract multi-level data analysis from intact glycopeptide data quantified by Glyco-Decipher.
Graphical Abstract
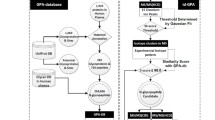
Brief process of GP-Marker

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glycosylation is a highly important post-translational modifications for proteins. It can be mainly categorized into N-glycosylation and O-glycosylation. In various diseases, alterations in the types and expression levels of glycosylation are often observed [1, 2]. The aberrant glycosylation levels have been found to be associated with the development of diseases such as autoimmune disorders [3], bacterial infections [4], viral infections [5], and cancer [6]. Therefore, investigating the differences in glycosylation between the health and disease can aid researchers in exploring potential early biomarkers for diseases.
With the advancements of the identification and quantification software tools, the sensitivity for identifying and quantifying of site-specific glycans by mass spectrometry has significantly improved [7,8,9,10]. In the typical workflow, the peptides carrying the intact glycans (abbreviated as intact glycopeptides) enriched from the digest of a proteome sample are subjected to LC–MS/MS and the acquired MS/MS spectra are then interpreted by software tools to identify the site-specific glycans. Different strategies have been developed to improve identification sensitivity [11]. For example, the Glyco-Decipher software developed by Fang et al. [7] uses the strategies of “Spectrum Expansion” to effectively improve the identification sensitivity of glycopeptides and uses the strategies of “Monosaccharide stepping” to assist in identifying the modified glycans that do not exist in the database. The identification results of intact glycopeptides include multi-level information such as glycans, glycosites, glycoproteins, and site-specific glycans. Traditionally, this information was obtained by using different omics techniques, i.e., glycomics to identify glycans released from glycoproteins and glycoproteomes to identify glycoproteins and glycosites from the deglycosylated proteins. Now, they are all presented in the spectra dataset of intact glycopeptides. However, manual analysis of the intact glycopeptide quantitative data at multiple levels is tedious and time consuming. The exploration of disease biomarkers often requires the analysis of a large amount of sample data acquired from a cohort of samples. For this case, convenient extraction of different levels of information on protein glycosylation at proteomics scale and find the characteristics and patterns of the data is critical.
As artificial intelligence technology continues to evolve, machine learning has demonstrated valuable application in the field of biological information processing [12]. J. Ochoteco Asensio et al. utilized information extracted from transcriptomics as feature to train machine learning model for imputing missing values in proteomics [13]. They reported that a random forest model demonstrated accurate predictions of proteins in new samples based on transcriptomics data. Sun et al. developed the Glycan Finder software; the software uses a deep learning model trained with a large amount of glycopeptide spectrum data to achieve de novo sequencing of glycans, thereby enabling the identification of unknown glycans [14].
Machine learning also has proven to be a powerful tool in the quest for disease biomarkers [15, 16]. Liu et al. developed a robust N-glycoproteomic platform that employs machine learning to identify four site-specific glycans as potential biomarkers for gastric cancer [17]. The platform demonstrates relatively accurate discrimination between healthy individuals and those with gastric cancer. Li et al. used the protein on small extracellular vesicles (sEVs) to train machine learning model and employed this model to subtype ovarian cancer, ultimately categorizing it into six distinct types with successful predictive outcomes [18]. Hence, machine learning is poised to become a common tool for uncovering potential features within datasets.
However, it is still very rare to extract different levels of information from the quantitative mass spectra dataset of intact glycopeptides and explore the potential of different level information, i.e., glycan, glycoprotein, glycosite, and site-specific glycan forms, for disease biomarker screening. Therefore, we hope to integrate convenient data processing methods and machine learning methods into one software for this purpose. The software should implement multi-level information extraction and machine learning processes for the processing of N-glycosylated intact glycopeptide data. Therefore, we have developed GP-Marker, a machine learning software that integrates six commonly used classification machine learning models. It can easily extract multi-level information from quantitative glycopeptide data quantified by Glyco-Decipher, and complete the entire process of missing value processing, T-test, and model training and prediction. The software also can output feature contribution files, allowing users to clearly understand the contribution of various features to classification. Additionally, it supports some plotting function to assist users in understanding data distribution and model training outcomes.
Method
GP-Marker code
The code was written using the PyCharm tool and python3.9 version. The software interface was written using version 6.5.3 of the Pyside6 package. Machine learning was written using version 1.3.2 of the Scikit-learn package. Other packages and versions are as follows: pandas (2.1.1); numpy (1.26.2); matplotlib (3.8.0). The executable program is freely available at https://github.com/DICP-1809.
Sample data
The intact glycopeptide dataset for demonstration of this software tool was from our published study [17]. The data was acquired as follows: (1) The intact N-glycopeptides were enriched by an automated hydrophilic interaction chromatography (HILIC) method from the tryptic digests of serum samples. In brief, the tryptic peptides were redissolved in 0.1% TFA/80% ACN and injected automatically onto a HILIC column at a flow rate of 200 μL min−1. After the glycopeptide fraction is collected, the next sample can be injected after the system is cleaned and re-equilibrated. The entire enrichment cycle for each sample is approximately 20 min. (2) The enriched glycopeptides were analyzed by a microflow LC–MS/MS system. In brief, the enriched glycopeptides derived from 5 μL serum were resuspended in 0.1% FA and analyzed in an Ultimate 3000 LC system connected online to an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific, USA). For shotgun glycoproteomes, DDA mode was operated to switch between MS and MS/MS acquisition. Glycopeptide fragmentation was performed by stepped HCD with normalized energy of 20%, 30%, and 40%. (3) The acquired HCD MS/MS spectra were interpretated by Glyco-Decipher software to identify and quantify the glycopeptides. In total, 200 samples from gastric cancer patients and health controls were analyzed. The discovery group included 70 serum samples from gastric cancer patients and 70 serum samples from healthy people, and the validation group included 30 serum samples from gastric cancer patients and 30 serum samples from healthy people.
Results
Overview of GP-Marker
The overall workflow of GP-Marker is shown in Fig. 1(a), and GP-Marker can be mainly divided into two functional modules (Fig. 1(b)): “missing value processing and T-test” and “Machine learning model training and prediction.”

Brief introduction to GP-Marker. (a) The workflow of GP-Marker for data processing. (b) The main interface of GP-Marker: (1) The blue box area corresponds to the missing value processing and T-test module. (2) The orange box area corresponds to the machine learning model training and prediction module
The missing value processing and T-test module (the blue part in Fig. 1(b)) is designed to (1) extract the quantitative information on four levels of protein glycosylation, i.e., the glycan level, the glycosite level, the protein level, and the site-specific glycans level, from the quantitative intact glycopeptide data between different groups and (2) statistically analyze the quantified glycosylation levels between different groups.
To obtain the quantitative information on different levels of protein glycosylation, we first extract the quantitative information of intact glycopeptides from the quantitative results outputted by Glyco-Decipher, and then, the quantitative value for each feature, e.g., a protein, a glycosite, a glycan, or a site-specific glycan, is determined by summing up the quantitative values of all intact glycopeptides from each feature. Taking protein level quantitative value determination as an example, we simply sum up the quantitative values of all glycopeptides derived from the same protein in each sample as the quantitative value of the corresponding protein in the corresponding sample. After the quantitative value extraction at each level is completed, statistical analysis can be performed.
For the statistical analysis, the missing value screening, missing value filling, and T-test to screen the significantly different features of the dataset are performed. Missing value screening is to filter out features with a certain number of missing values in the data. GP-Marker provides three missing value screening methods, which can obtain screening results with different screening stringencies. GP-Marker provides some missing value filling methods, including directly filling in zero, filling in the median of data, and filling in random values according to the left-skewed peak of the normal distribution of the original dataset. Filling random values with the left-skewed peak [19, 20] of the normal distribution means fitting all the data in the dataset into a normal distribution and then adjusting its mean and variance so that the new normal distribution peak is located on the left side of the original normal distribution. In this way, the random values generated through the new normal distribution are all smaller values in the original normal distribution. Since the missing values in the mass spectrometry data are likely to be unable to be identified due to low abundance, this method of using a lower random value to fill the missing values may be a situation more like the real situation. After completing the missing value processing, T-test can be performed to select significantly different features. After classification, GP-Marker can perform T-test on each feature. Users can select the corresponding P-value and fold change to filter features and generate the input files required for machine learning.
The machine learning model training and prediction module is designed to facilitate the screening of feature candidates for the classification of two different groups of samples (the orange part in Fig. 1(b)), GP-Marker provides functions such as model training, optimization, contribution output, and prediction. The model training function provides six classic classification models for users to choose, namely logistic regression [21], random forest, decision tree, support vector machine, naive Bayes, and K nearest neighbor. Different models have differences in feature recognition, and users can try multiple models to choose the model that suits their own dataset. Model training uses the most basic model parameters. If the user thinks that the basic model parameters are inappropriate, GP-Marker also provides a grid search method to automatically optimize the model parameters. For the evaluation of the quality of the model, prediction accuracy information will be output after the model is trained, and users can also use the cross-validation method to view multiple training situations. In addition, receiver operating characteristic (ROC) curve plotting and learning curve plotting of the model are also provided to provide an intuitive display of model performance. After completing model training, users can directly export the feature contribution file, which will give the contribution of each feature to provide information on feature importance. At the same time, GP-Marker can directly output the area under the ROC curve (AUC) calculated based on the data for each candidate biomarker feature. Combined with the contribution ranking, features with higher rankings and larger AUC values are expected to be promising disease biomarkers. By using GP-Marker, potential biomarkers of protein glycosylation at different levels, i.e., glycoprotein, glycosite, glycan, and site-specific glycans, could be easily obtained from the quantitative dataset of intact glycopeptides acquired from a large cohort of sample.
GP-Marker allows the analysis and display of intact glycopeptide quantitative data at different levels
The quantitative intact glycopeptide datasets containing four levels of information, i.e., glycans, glycosite, protein, and site-specific glycans. GP-Marker allows statistics analysis of intact glycopeptide datasets and reports the significantly changed feature at different levels. A common differential analysis method is generally to compare the quantitative differences between health and disease for a large cohort of samples. At each level, T-test analysis on each feature is performed to find features with significant changes. GP-Marker integrates all process of traditional methods and provides a series of visualization methods to visually display the processing results, such as volcano plots, box distribution plots, principal component analysis (PCA), and ROC curves. The boxplots, volcano plots, PCA, and ROC curves below are all drawn directly by GP-Marker. Here, a quantitative intact glycopeptide dataset (Supplementary Data 1) acquired from the tryptic digest of serum samples with a sample size of 70 healthy and 70 gastric cancer patients [17] is used to demonstrate the statistical analysis method in GP-Marker at four levels of glycans, glycosite, glycoprotein, and site-specific glycans. In order to have an overall understanding of the data, we analyze it from the aspects of the difference in data distribution and the significance of feature changes. First, we counted the number of identifications in health and disease at each level and plotted them into boxplots (Fig. 2). From the boxplot, we can find that at each level, the number of identifications in the disease group is greater than that in the healthy group, indicating that at each level, the disease group has higher heterogeneity. This result may indicate that the glycosylation modification process becomes more complex and variable during the development of the disease, resulting in more abundant glycosylation modifications. This change may not only be a change in the glycan but also bring about a change in the glycosylation modification site or protein. This is also consistent with our understanding of micro-heterogeneity and macro-heterogeneity in glycosylation.

GP-Marker enables direct plotting of boxplots for the number of unique identifications at different levels of protein glycosylation. (a) ~ (d) glycan, protein, glycosite, and site-specific glycan (TD: disease data; CD: health data; Dataset: the discovery group included 70 serum samples from gastric cancer patients and 70 serum samples from healthy people)
Next, after removing features with missing value ratios of both health and disease greater than 0.3, we conducted a T-test analysis on the data at each level and drew a volcano plot (Fig. 3) to find the features with significant changes in each level. We conduct a T-test on each level, and then select features with the following criteria: P-value less than 0.01 and fold change greater than 2 or less than − 2 as features with significant differences, and then draw a volcano chart according to this criteria. From the T-test results (Supplementary Data 2), we screened out 11 glycans, 8 proteins, 24 glycosites, and 154 site-specific glycans that pass the criteria. The proportions of each level that pass the criteria are 8%, 7.8%, 10.6%, and 16.7% respectively. These data indicate that changes caused by disease are more obvious at the glycosite and site-specific glycan level than at the protein level and glycan level. This may be because the data at other levels are accumulated from the data at the site-specific glycan level, causing other levels to lose some information compared to the site-specific glycan level, resulting in less variability. This is also a limitation of this statistical method. It can only provide a rough comparison, rather than a very accurate quantification. It can be seen from the volcano diagrams at each level that the number of features that pass the criteria is relatively large. However, our disease biomarker screening often focuses on one or a combination of several features. Too many of features obviously cannot meet the screening needs. Therefore, we need to use other methods to narrow down the scope of candidate markers, such as machine learning and calculating AUC values.

GP-Marker enables direct plotting of volcano diagram at different level of protein glycosylation. (a) ~ (d) glycan, protein, glycosite, and site-specific glycan. Red dots are significant upward features, blue dots are significant downward features (Dataset: the discovery group included 70 serum samples from gastric cancer patients and 70 serum samples from healthy people)
Then, we retain features that pass the T-test criteria and perform principal component analysis (PCA) on the data at each level to obtain the distribution of healthy and disease sample data at each level. In Fig. 4, the overlap of healthy and disease sample data at the glycan and protein levels is more than that at the site and site-specific glycan levels. This suggests that distinguishing healthy and disease sample data is more difficult at the glycan and protein levels than at the site and site-specific glycan levels. This is also consistent with the results of the volcano plot. Therefore, through PCA, we can also preliminarily judge at which levels it may be better to find biomarkers.

GP-Marker enables direct plotting of principal component analysis (PCA) at different levels of protein glycosylation. (a) ~ (d) glycan, protein, glycosite, and site-specific glycan. Gray dots are healthy samples, blue dots are disease samples (Dataset: the discovery group included 70 serum samples from gastric cancer patients and 70 serum samples from healthy people)
Through above statistical analysis of the data, we can preliminarily understand the differences between healthy and disease samples in the data, laying the foundation for our further processing and exploration. At the same time, in the above statistical analysis, the number of features screened out is often relatively large, which cannot meet the requirements of disease biomarkers screenings. Therefore, we need to use some other methods to find one or several features with the largest difference or better discriminating effect. The development of artificial intelligence and its application in life sciences has brought us some great tools, such as machine learning.
GP-Marker facilitates the screening of Glyco-Biomarker candidates at different levels
A disease biomarker should be able to accurately distinguish disease and healthy samples. While ensuring good diagnostic performance, disease biomarkers should be one or more features that are relatively simple and easy to detect. Simply using statistical analysis mainly reflects the average level of features and cannot focus on the changes in each sample. Moreover, the number of significantly different features obtained may be large, which cannot meet the requirements for disease biomarker screening. Therefore, we can train supervised machine learning models with features that have been statistically analyzed to obtain the contribution of each feature in distinguishing between diseased and healthy samples, thus providing guidance for disease biomarker screening. GP-Marker can easily implement the entire process of machine learning biomarker screening. Hence, machine learning screening was performed at the four levels of glycans, glycosite, glycoprotein, and site-specific glycans for the gastric cancer glycopeptide dataset, and the discrimination performance of biomarkers at different levels was compared.
The operation processes of machine learning at different levels are basically the same, the only difference is the dataset input to the machine learning model. Here, we take the protein level data as an example. First, import the protein level dataset into GP-Marker, then divide the samples into disease and health categories, retain features with a missing value ratio less than 0.3 in a certain category, and replace the remaining missing values with 0. This operation preserves features that can be quantified in most samples. Then perform T-test to calculate each feature’s P-value and fold change in the two categories, and then retain the P-value greater than 2 (− log10 P-value), and the fold change greater than 1, or fold change less than − 1 (log2 Fold Change). These features are imported into the random forest model for training, and the accuracy of the training set and the contribution of each feature can be obtained. Finally, the validation set (Supplementary Data 3) can be imported to obtain the prediction accuracy information of the validation set. The operation process for other levels is the same, except that the starting dataset needs to be replaced with the dataset of this level. Finally, features with higher contribution and larger AUC value are the candidates for disease biomarkers (Fig. 5).

GP-Marker facilitates the screening of Glyco-Biomarker candidates at different levels. (a) Feature contribution ranking and AUC value joint screening strategy, and ROC curve of Q06033. (b) The ROC of Hex(7)HexNAc(6)NeuAc(4)Fuc(1). (c) The ROC of P04114@2982. (d) The ROC of Hex(5)HexNAc(4)NeuAc(2)P04114@2982. (e) P04114 boxplots in health and disease. (f) P04114@2982 boxplots in health and disease (TD: disease data; CD: health data; Dataset: the discovery group included 70 serum samples from gastric cancer patients and 70 serum samples from healthy people)
After completing the machine learning model training, GP-Marker can output the feature contribution file (Supplementary Data 4) as well as the AUC value for each feature (Supplementary Data 5). We can identify candidates for disease biomarkers based on the feature contribution ranking and the AUC value of the feature. Taking the protein level as an example (Fig. 5(a)), the protein ranked first in the contribution file is ITIH3 (Q06033) and its AUC value is also the largest; we can consider this protein has the potential candidate to be used as a disease biomarker. The screening procedure at other levels is the same; of course, the results of feature contribution ranking and AUC will not be consistent as in the protein level. We can combine various information to determine the screening of disease biomarkers. The corresponding relationship between feature contributions and AUC values at other levels is shown in the Supplementary Information (Supplementary Fig. 1). It is worth noting that a feature with a higher contribution can only mean that it has a larger weight among all the features of the training model, but it does not mean that using this feature alone can also achieve better discrimination.
Here, we use the same workflow to analyze the data at four levels. We first screened the top five features according to the ranking of feature contribution, then calculated the AUC values, and selected the features with the highest AUC value as the representative differential feature at each level of screening.
As shown in Fig. 5(a) ~ (d), the representative differential features are as follows: the glycan level is Hex(7)HexNAc(6)NeuAc(4)Fuc(1), the site level is P04114@2982, the protein level is ITIH3 (Q06033), and the site-specific glycan level is Hex(5)HexNAc(4)NeuAc(2)P04114@2982. The ROC curve and AUC value of each feature are calculated from its quantitative data by using the Scikit-learn package (Supplementary Data 5).
The site is consistent with the site-specific glycan, and this site-specific glycan is also one of the 18 site-specific glycans screened by Liu et al. [17]. We found the glycans contain sialic acid or fucose, which is consistent with the fact that these glycans in human serum proteins are closely related to the development of the disease [22]. We also found that the site level and site-specific glycan level are consistent, while the glycan and protein levels are quite different. Therefore, we examined protein APOB (P04114) and glycan (Hex(5)HexNAc(4)NeuAc(2)) distribution (Supplementary Fig. 2) among site-specific glycans. It can be seen from the box distribution plot (Fig. 5(e)) that the protein has no major difference in data distribution between the healthy and disease samples. This may be because that the overall abundance of the protein does not change significantly in health and disease, but the sites on it are both upregulated and downregulated in samples, and the distribution difference of P04114@2982 in health and disease is larger than that of APOB (P04114) (Fig. 5(f)). Through this inspection, we can also attribute the differences to specific level changes.
Through the AUC value of a single feature, we can obtain the diagnostic effect of each feature in distinguishing healthy and disease samples. However, sometimes multiple features may be combined to improve diagnostic accuracy. Currently, it is necessary to examine the results of machine learning training for using multiple features. We refer to the screening strategy of Liu et al. [17], and at each level, select the top four feature combinations with the highest feature contribution to retrain the machine learning model (Supplementary Data 6), and demonstrate its diagnostic effect in distinguishing healthy and disease samples.
From the accuracy of each level (Supplementary Fig. 3), we can find that the discrimination effect at the site and site-specific glycan levels is better than that at the glycan and protein levels in discovery dataset, which is also consistent with the PCA results in the second part of this study. This result may be due to the lack of more detailed information at the glycan and protein levels compared to the site and site-specific glycan levels, resulting in many differences being ignored. However, in the validation dataset, the accuracy of the combination of four site-specific glycans we selected has a sudden drop, which may be due to overfitting of the trained model. The combination of the four site-specific glycans screened by Liu et al. [17] still performed well, which shows that machine learning can only be used as a tool to help us find some possible candidates, and it is necessary to combine experimental conditions and biological information for final screening (Supplementary Fig. 4). However, we can provide glycosite, glycoprotein, and glycan level information in addition to site-specific glycans to help us understand the data from multiple perspectives. This result may indicate that gastric cancer does not have a significant impact on the abundance of overall proteins and glycans in serum but has a greater impact on specific glycosylation modifications.
Through GP-Marker training and analysis of four levels of machine learning models, the diagnostic effect of each level can be obtained. At the same time, by comparing the screening results at each level, we can find some proteins with both upregulated and downregulated sites, which can help us attribute the differences caused by the disease to specific levels. This can also provide some new clues for the screening of disease biomarkers related to protein glycosylation.
Discussion
Different software tools including pGlyco3 [23], StrcucGP [10], Glyco-Decipher [7], and Glyco-Finder [14] are developed to interpretate the MS/MS spectra of intact N-glycopeptides enriched from protein digest, which can yield the identification of the glycopeptides. Some of these tools as well as some specialized tools, e.g., pGlycoQuant [24], could perform quantitative analysis of intact N-glycopeptides between samples. Each intact N-glycopeptide contains four level of information, i.e., glycan, glycoprotein, glycosite, and site-specific glycans. The above software tools can only be used to identify and quantify glycopeptides, but do not offer the function to analyze protein glycosylation at different levels. In this study, we developed a software tool called GP-Marker to facilitate large-scale data mining of quantitative spectra dataset of intact glycopeptide at multiple levels. The quantitative information on different levels, i.e., glycan, glycoprotein, glycosite, and site-specific glycans, could be easily extracted, analyzed, and visualized. The potential of these different level information for biomarker discovery could also be easily evaluated. It can realize the entire process from machine learning model training to prediction, including missing value screening, fill in missing value, data normalization, T-test, and model training and prediction. After completing the model training, the contribution of each feature to the model can be output to illustrate the importance of features in distinguishing samples.
At the same time, to allow users to understand the data distribution, model training, and the significance of the output features more intuitively, GP-Marker also provides a variety of plotting methods. These include boxplots and pie charts for understanding data distribution, volcano plot and PCA plots for assessing sample differences, ROC plots and learning curve for evaluating model training, ROC curves for an individual feature on the dataset, and bar charts and heatmaps for visualizing the distribution of specific glycan structures. Through the series of visualizations, users can gain a comprehensive understanding of the features of the quantitative mass spectra dataset of intact glycopeptides.
We use GP-Marker to screen biomarkers for gastric cancer datasets we acquired before at four levels: glycan, glycoprotein, glycosite, and site-specific glycans. GP-Marker can get the accuracy of prediction at each level, and we can get the contribution of features that provides a reference for our further biomarker screening. At the same time, the feature differences we directly screen out at different levels can help us focus the abundance differences caused by the disease to specific levels and can also help us understand the specific impact of the disease on each level to a certain extent.
It should be noted that GP-Marker is not a software tool for interpretation of glycopeptide MS/MS spectra or quantification of glycopeptides. It aims to extract and process four levels of information, i.e., glycan, glycoprotein, glycosite, and site-specific glycan, from the quantitative N-linked intact glycopeptide dataset with intact glycopeptides already identified and quantified. The determination of glycosite is indispensable for the determination of glycan and site-specific glycan. However, the determination of O-glycosites on O-linked intact glycopeptides is often difficult because O-glycosylation often has multiple sites on a peptide. Therefore, GP-Marker is currently unable to perform multi-level processing of O-glycosylation data analysis.
Conclusion
In this study, we presented a software tool called GP-Marker to facilitate the analysis of the quantitative dataset of N-linked intact glycopeptides derived from protein digest. GP-Marker can provide glycosite, glycoprotein, and glycan level information in addition to site-specific glycans to help us understand the data from multiple perspectives. GP-Marker also provides a variety of plotting methods and can realize the entire process from machine learning model training to prediction, which can help us to screening the biomarker for each level. We believe that the user-friendly tool is very helpful for biological research on disease biomarker screening of glycoproteins.
Data availability
GP-Marker is available at https://github.com/DICP-1809.
References
Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015. https://doi.org/10.1038/nrc3982.
Wang M, Zhu J, Lubman DM, Gao C. Aberrant glycosylation and cancer biomarker discovery: a promising and thorny journey. Clin Chem Lab Med. 2018. https://doi.org/10.1515/cclm-2018-0379.
Ohmi Y, Ise W, Harazono A, Takakura D, Fukuyama H, Baba Y, Narazaki M, Shoda H, Takahashi N, Ohkawa Y, Ji S, Sugiyama F, Fujio K, Kumanogoh A, Yamamoto K, Kawasaki N, Kurosaki T, Takahashi Y, Furukawa K. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat Commun. 2016. https://doi.org/10.1038/ncomms11205.
Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, Schoen MK, Tafesse F, Martin C, Leung V, Mahan AE, Sips M, Kumar MP, Tedesco J, Robinson H, Tkachenko E, Draghi M, Freedberg KJ, Streeck H, Suscovich TJ, Lauffenburger DA, Restrepo BI, Day C, Fortune SM, Alter G. A functional role for antibodies in tuberculosis. Cell. 2016. https://doi.org/10.1016/j.cell.2016.08.072.
Byrd-Leotis L, Liu R, Bradley KC, Lasanajak Y, Cummings SF, Song X, Heimburg-Molinaro J, Galloway SE, Culhane MR, Smith DF, Steinhauer DA, Cummings RD. Shotgun glycomics of pig lung identifies natural endogenous receptors for influenza viruses. Proc Natl Acad Sci USA. 2014. https://doi.org/10.1073/pnas.1323162111.
Kailemia MJ, Park D, Lebrilla CB. Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem. 2016. https://doi.org/10.1007/s00216-016-9880-6.
Fang Z, Qin H, Mao J, Wang Z, Zhang N, Wang Y, Liu L, Nie Y, Dong M, Ye M. Glyco-Decipher enables glycan database-independent peptide matching and in-depth characterization of site-specific N-glycosylation. Nat Commun. 2022. https://doi.org/10.1038/s41467-022-29530-y.
Cho BG, Gutierrez Reyes CD, Goli M, Gautam S, Banazadeh A, Mechref Y. Targeted N-glycan analysis with parallel reaction monitoring using a quadrupole-orbitrap hybrid mass spectrometer. Anal Chem. 2022. https://doi.org/10.1021/acs.analchem.2c01975.
Messner CB, Demichev V, Bloomfield N, Yu JSL, White M, Kreidl M, Egger A-S, Freiwald A, Ivosev G, Wasim F, Zelezniak A, Jürgens L, Suttorp N, Sander LE, Kurth F, Lilley KS, Mülleder M, Tate S, Ralser M. Ultra-fast proteomics with Scanning SWATH. Nat Biotechnol. 2021. https://doi.org/10.1038/s41587-021-00860-4.
Shen J, Jia L, Dang L, Su Y, Zhang J, Xu Y, Zhu B, Chen Z, Wu J, Lan R, Hao Z, Ma C, Zhao T, Gao N, Bai J, Zhi Y, Li J, Zhang J, Sun S. StrucGP: de novo structural sequencing of site-specific N-glycan on glycoproteins using a modularization strategy. Nat Methods. 2021. https://doi.org/10.1038/s41592-021-01209-0.
Cao W. Advancing mass spectrometry–based glycoproteomic software tools for comprehensive site-specific glycoproteome analysis. Curr Opin Chem Biol. 2024. https://doi.org/10.1016/j.cbpa.2024.102442.
Cox J. Prediction of peptide mass spectral libraries with machine learning. Nat Biotechnol. 2022. https://doi.org/10.1038/s41587-022-01424-w.
OchotecoAsensio J, Verheijen M, Caiment F. Predicting missing proteomics values using machine learning: filling the gap using transcriptomics and other biological features. Comput Struct Biotechnol J. 2022. https://doi.org/10.1016/j.csbj.2022.04.017.
Sun W, Zhang Q, Zhang X, Tran NH, ZiaurRahman M, Chen Z, Peng C, Ma J, Li M, Xin L, Shan B. Glycopeptide database search and de novo sequencing with PEAKS GlycanFinder enable highly sensitive glycoproteomics. Nat Commun. 2023;14:4046. https://doi.org/10.1038/s41467-023-39699-5.
Dong M, Lih TM, Chen S-Y, Cho K-C, Eguez RV, Höti N, Zhou Y, Yang W, Mangold L, Chan DW, Zhang Z, Sokoll LJ, Partin A, Zhang H. Urinary glycoproteins associated with aggressive prostate cancer. Theranostics. 2020;10:11892–907. https://doi.org/10.7150/thno.47066.
Lih T-SM, Dong M, Mangold L, Partin A, Zhang H. Urinary marker panels for aggressive prostate cancer detection. Sci Rep. 2022;12:14837. https://doi.org/10.1038/s41598-022-19134-3.
Liu L, Liu L, Wang Y, Fang Z, Bian Y, Zhang W, Wang Z, Gao X, Zhao C, Tian M, Liu X, Qin H, Guo Z, Liang X, Dong M, Nie Y, Ye M. Robust glycoproteomics platform reveals a tetra-antennary site-specific glycan capping with Sialyl-Lewis antigen for early detection of gastric cancer. Adv Sci. 2023. https://doi.org/10.1002/advs.202306955.
Li J, Li Y, Qin Li Lu, Sun QT, Zheng L, Ye Lu, Zhu J, Fengli Qu, Tan W. An aptamer-based nanoflow cytometry method for the molecular detection and classification of ovarian cancers through profiling of tumor markers on small extracellular vesicles. Angew Chem Int Ed. 2023. https://doi.org/10.1002/anie.202314262.
Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016. https://doi.org/10.1038/nmeth.3901.
Jin L, Bi Y, Hu C, Qu J, Shen S, Wang X, Tian Y. A comparative study of evaluating missing value imputation methods in label-free proteomics. Sci Rep. 2021;11:1760. https://doi.org/10.1038/s41598-021-81279-4.
Liu X-Y, Wu S-B, Zeng W-Q, Yuan Z-J, Xu H-B. LogSum + L 2 penalized logistic regression model for biomarker selection and cancer classification. Sci Rep. 2020. https://doi.org/10.1038/s41598-020-79028-0.
Zhang Z, Wuhrer M, Holst S. Serum sialylation changes in cancer. Glycoconj J. 2018. https://doi.org/10.1007/s10719-018-9820-0.
Zeng W-F, Cao W-Q, Liu M-Q, He S-M, Yang P-Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. 2021. Nat Methods. https://doi.org/10.1038/s41592-021-01306-0.
Kong S, Gong P, Zeng W-F, Jiang B, Hou X, Zhang Y, Zhao H, Liu M, Yan G, Zhou X, Qiao X, Wu M, Yang P, Liu C, Cao W. pGlycoQuant with a deep residual network for quantitative glycoproteomics at intact glycopeptide level. 2022. Nat Commun. https://doi.org/10.1038/s41467-022-35172-x.
Funding
This work was supported, in part, by funds from the National Key Research and Development Program of China (2022YFC3400801, 2021YFA1302601), the National Natural Science Foundation of China (22034007, 22274014, 22222409, 92253301), and the innovation program of science and research from the DICP, CAS (DMU-2&DICP UN202308).
Author information
Authors and Affiliations
Contributions
Qi Liu: conceptualization, software, visualization, writing original draft. He Zhu: discussion and assistance. Zheng Fang: discussion and assistance. Mingming Dong: supervision. Hongqiang Qin: supervision. Mingliang Ye: project administration, funding acquisition, supervision, writing reviewing and editing. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection featuring Current Progress in Glycosciences and Glycobioinformatics with guest editors Joseph Zaia and Kiyoko F. Aoki-Kinoshita.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Q., Zhu, H., Fang, Z. et al. GP-Marker facilitates the analysis of intact glycopeptide quantitative data at different levels. Anal Bioanal Chem (2024). https://doi.org/10.1007/s00216-024-05499-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00216-024-05499-z