
Abstract
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is used routinely in clinical diagnostics; however, automating the sample pretreatment is challenging. We established and evaluated an automated method based on the magnetic bead extraction principle (MBE) to measure normetanephrine (NMN), metanephrine (MN), and 3-methoxytyramine (3-MT). The target analytes were extracted, purified, and concentrated using different solvents and chemical bond–modified magnetic beads transferred via a magnetic bar. The linearity, recovery, matrix effect, and precision of MBE were evaluated thoroughly, and compared with traditional solid-phase extraction (SPE) using 131 plasma samples. The chromatography peaks of metanephrines and 3-MT, extracted via MBE, are symmetrical, without interfering peaks. The linearity was excellent with correlation coefficient (r) > 0.99. The MBE exhibited good reproducibility with within-run coefficient variations (CVs) of 1.96–2.00%, 4.06–5.75%, and 3.89–4.90% for MN, NMN, and 3-MT, respectively. The total CVs for MN, NMN, and 3-MT were 1.96–2.80%, 5.12–5.75%, and 5.44–6.27%, respectively. The relative recoveries for MN, NMN, and 3-MT varied between 93.5 and 107.4%, whereas their biases were all within 10%. The results for MN, NMN, and 3-MT extracted via MBE compared with SPE exhibited excellent correlation, with r > 0.99; the mean bias% for MN, NMN, and 3-MT were small (−2.9%, −3.2%, and −3.2%, respectively). In conclusion, the automated MBE method for measuring plasma metanephrines and 3-MT can be applied in future routine clinical diagnostics, and the MBE principle may indicate a new era for LC-MS/MS in clinical application.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liquid chromatography tandem mass spectrometry (LC-MS/MS) has been used in clinical laboratories since the 1980s. It has the advantage of sensitivity and specificity and has attracted significant attention in clinical practice. LC-MS/MS can measure biomarkers that already have immunoassay methods but are non-specific and exhibit cross-reactivity or low sensitivity [1, 2], and can measure biomarkers that did not previously have mature immunoassay or biochemical methods, such as metanephrines [3, 4] and amino acids [5]. Recently, some clinical guidelines have recommended LC-MS/MS as the initial choice for measuring metanephrines [6].
Although laboratory staff members acknowledge that LC-MS/MS has several advantages and has the potential to provide more accurate results for clinicians [7], it has been used mostly for research, and only a few clinical laboratories use LC-MS/MS in real clinical practice. LC-MS/MS is not as widely used in clinical laboratories as the automatic biochemical or immunoassay stream-lined analyzers in the intelligent clinical laboratory era. There are two reasons which might account for the limited clinical application of LC-MS/MS: it is expensive, and there are few skilled technicians who are familiar with this method. However, the most important reason may be the complicated serum or urine pretreatment procedures required before LC-MS/MS analysis. The most used pretreatment methods for LC-MS/MS are liquid-liquid extraction (LLE), solid-phase extraction (SPE), and derivatization procedures [8, 9]. For the measurement of metanephrines or steroids, complicated manual pretreatment steps with SPE [10] are usually required. Although partially automated methods have been developed by refitting automated enzyme-linked immunosorbent assay workstations for LLE or SPE [8], LC-MS/MS involves several steps, centrifugation and positive pressure operation are often needed, and it is time-consuming. With the progress of LC-MS/MS in clinical laboratories, fully automated pretreatment methods are needed to ensure wide usage in clinical practice, especially for busy hospitals. Herein, we develop and validate an automated magnetic bead extraction (MBE) method that mimics SPE, but is much simpler. The magnetic beads were modified with porous copolymer using lipophilic divinylbenzene and hydrophilic N-vinylpyrrolidone similar to the traditional hydrophile-lipophile balance plate (HLB plate) sorbent instead of the antibodies used in immunoassays. A magnetic bar transfers the beads, and the target analytes absorbed on the magnetic beads are extracted, purified, and concentrated. This method can be used to measure compounds such as metanephrines, steroids, and vitamin D. Considering that LC-MS/MS is the recommended choice for measuring plasma metanephrines (which play key roles in the diagnosis of pheochromocytoma and paraganglioma (PPGLs) [6, 11]), and has a clinical sensitivity and specificity > 90%, we validated the MBE method and compared it with the SPE method used in our laboratory for measuring plasma metanephrines and 3-methoxytyramine.
Experimental section
Materials and plasma samples
The SPE method used metanephrine hydrochloride (1045±1μg/mL, 98.93%), normetanephrine hydrochloride (1042±1μg/mL, 99.3%), and 3-methoxytyramine hydrochloride (1072±1μg/mL, 99.26%) purchased from IsoScience (King of Prussia, PA, USA) as calibrators, and metanephrine-d3 hydrochloride (MN-d3, 98.64%), normetanephrine-d3 hydrochloride (NMN-d3, 99.76%), and 3-methoxytyramine-d4 hydrochloride (3-MT-d4, 98.63%) from IsoScience as their isotope-labeled internal standards (IS). HPLC-grade solvents including acetonitrile and methanol were obtained from Fisher Scientific (Pittsburgh, PA, USA). HPLC-grade formic acid was purchased from Sigma-Aldrich (St. Louis, MO, USA). A MilliQ Advantage ultrapure water system (Millipore, MA, USA) was used to produce ultrapure water. The MBE pretreatment kit was provided by GBI (Beijing BGI-GBI Biotech Co., Ltd, Beijing, China.). The plasma for the method validation and comparison was obtained from that remaining of patient samples in the Department of Laboratory Medicine, Peking Union Medical College Hospital. All the procedures performed in the studies involving human participants were in accordance with the ethical standards of the Ethics Committee of Peking Union Medical College & Chinese Academy of Medical Sciences, Peking Union Medical College Hospital (JS-2956).
Preparation of stock solutions and calibration curves
A stock solution with a concentration of 2262.5 nmol/L, 2385.5 nmol/L, and 2571.0 nmol/L for MN, NMN, and 3-MT, respectively, was prepared by transferring the standards and methanol (0.1% ascorbic acid) into a volumetric flask (Class A). The working calibrators with the highest concentrations of 45.2 nmol/L, 47.71 nmol/L, and 51.42 nmol/L were prepared by diluting the stock solution. A combined IS stock solution with a concentration of 5 ng/mL of MN-d3, NMN-d3, and 3-MT-d4 was prepared in methanol containing 0.1% ascorbic acid. The combined internal standard stock solution was directly diluted 100 times using ultrapure water before analysis to obtain a working IS solution. The stock solution of the mixed standards and IS was aliquoted in amber tubes (1 mL) and stored at −80 °C until analysis.
Pretreatment method
MBE pretreatment method
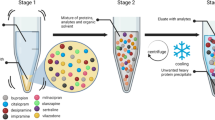
The magnetic beads used were modified with porous copolymer polymerized using lipophilic divinylbenzene and hydrophilic N-vinylpyrrolidone to form an HLB characteristic (Fig. 1). The MBE pretreatment method was performed on the automated extraction instrument GBI EX-48 (Beijing BGI-GBI Biotech Co., Ltd, Beijing, China). The calibrators (200 μL), quality control or serum, and ISs (200 μL) were added into line 3 or 9 on the 96-deep well plate pre-filled with extraction beads and solvents, and the following steps including condition, equilibrium, wash, and elution were performed automatically on the GBI EX-48 (Fig. 1).

Principles and steps involved in the magnetic bead extraction method. Conditioning solvents (5 mg/mL magnetic beads in 50% methanol-water), equilibrium solvents, sample + IS, weak wash solvents, strong wash solvents, and eluting solvents were placed in vials of lines 1(7), 2(8), 3(9), 4(10), 5(11), 6(12) of the 96-well microplate, respectively
SPE pretreatment method
The SPE pretreatment method was performed according to our previously published article [10]. The sample (200 μL, plasma, quality control, or calibrators) was accurately transferred into a glass tube (5 mL) using a micropipette, followed by the addition of the working IS solution (200 μL), and vortexed for 1 min. A 350-μL volume was transferred into a pre-conditioned 96-well SPE plate, which was then loaded and washed with pure water (200 μL) followed by pure acetonitrile (200 μL). The samples were then eluted with acetonitrile (60 μL, 85% with 1% formic acid) on the SPE Positive Pressure-96 processor and collected using a 700-μL round 96-well sample plate. The eluates were dried under nitrogen gas for 10 min, reconstituted with water (40 μL), and analyzed using the LC-MS/MS system.
LC-MS/MS instrumentation parameters
The LC-MS/MS measurement was performed on a Waters ACQUITY UPLC I-Class in tandem with a Waters Xevo TQ-S mass spectrometry system. The gradient elution program and MS/MS detection conditions were the same as those previously published [10]. The mobile phase consisted of 0.1% formic acid in water as phase A, and acetonitrile as phase B. An HSS PFP column (2.1 × 100 mm × 1.7 μm) (Waters Corporation, Milford, MA, USA) was used. The gradient elution program was as follows: 0–1 min, 98% A; 1–2.5 min, 98 to 90% A; 2.5–3 min, 90 to 10% A; 3.0–3.5 min, 10% A; and 3.5–4.0 min, 10 to 98% A; the flow rate was 0.4 mL/min. MS/MS detection was performed using positive electrospray ionization using multiple reaction modes (MRM). The MRM transitions for each compound were as follows: 180.0→148.1 (MN, quantifier), 180.0quanti (MN, qualifier), 183.0 151.0 (MN-IS), 166.1er), r each compound were as follows: 180.0→148.1s (MRM). in tandem with a Wa151.1→119.1 (3-MT, quantifier), 151.1→91.0 (3-MT, qualifier), and 155.0→95.0 (3-MT-IS, 155→123 can also be sued). The source temperature was 150 °C and the desolvation gas temperature was 600 °C; the capillary voltage was 0.5 kV, and the desolvation gas flow rate was 1100 L/h.
Comparison between MBE and SPE methods
The samples (130) were collected from routine metanephrine measurement requests in our clinical laboratory at the Peking Union Medical College Hospital. Each sample was divided into two tubes and stored at −80 °C; the aliquots were pretreated simultaneously using the SPE and MBE methods by two experienced operators, and then analyzed using the same LC-MS/MS instrument (the Waters TQ-S). Regression analysis and Bland-Altman plots were used for comparing the MBE and SPE methods. The Bland-Altman plots were used to identify the mean bias (the average of the difference between the measurements obtained from the two assays was also compared).
Statistics
Statistical analyses were performed using Microsoft Excel 2010 and the MedCalc Statistical Software. Method comparison was performed using Passing-Bablok regression and Bland-Altman plots. A p < 0.05 was considered statistically significant.
Results
Layout and process of the MBE method
The vials of lines 1(7), 2(8), 3(9), 4(10), 5(11), and 6(12) of the 96-deep well plate were designated for placing magnetic beads, equilibrium solvents, samples + IS, weak wash solvents, strong wash solvents, and eluting solvents, respectively. After the samples and IS were added to the vials, the extraction process was performed using a magnetic rod to transfer the magnetic beads. The magnetic rods were covered by disposable protective stirrer sheaths, and were then placed in line 1 on the 96-deep well plate pre-fitted with extraction magnetic beads, stirred for 1 min to ensure that the magnetic beads were conditioned and well distributed; the magnetic beads were then transferred into line 2, and stirred for 1 min to equilibrate the magnetic beads; the magnetic beads were transferred into line 3 where the samples and IS were located and stirred for 5 min to extract the target analytes. Next, the magnetic beads were transferred sequentially into lines 4 and 5 and stirred for 30 s to remove any interference and impurities. The magnetic beads were then transferred into line 6, stirred for 1 min to further elute the target analytes. The total extraction time of one batch was 10 min. The general steps and principle are presented in Fig. 1.
The conditioning, washing, and eluting solvents of the MBE method were similar to those used for the SPE process; however, unlike the SPE process, where the target compounds were extracted onto the solid phase that is fixed on the tube, in the MBE process, the target compounds were extracted onto the magnetic beads and were transferred into each solvent for purification and concentration. The process does not need a positive pressure process and instrument; thus, it is simpler to realize full automation, similar to the completely automatic biochemical or immunoassay analyzer.
Representative chromatography of the MBE method
After the extraction of the plasma samples, the eluates were dried under nitrogen, reconstituted, and then analyzed using the LC-MS/MS. A representative chromatogram of metanephrines extracted from a plasma sample is illustrated in Fig. 2. Compared with the SPE extraction method used previously in our hospital, the extract from the MBE method does not contain additional interfering or miscellaneous peaks. The signal-to-noise is similar in the two methods (Supplemental Figures 1-2).

Representative chromatogram of extract from the MBE method. From top to bottom, chromatograms are for MN, IS of MN, NMN, IS of NMN, 3-MT, and IS of 3-MT, respectively
Linearity, limit of detection (LOD), and limit of quantification (LOQ) of the MBE method
According to CLSI-C62a [12], at least six to eight no-zero calibrators are needed to form the final calibrators. In this study, nine levels of calibrators were used to generate the calibration curves for the MBE method. For the ranges of 0.023–45.25 nmol/L (MN), 0.048–47.71 nmol/L (NMN), and 0.01–25.71 nmol/L, with quadratic polynomial regression weighted by 1/x, the linearity exhibited excellent correlation (correlation coefficient, (r) > 0.99) in all batches for the three compounds. A representative linear regression is presented in Supplemental Figs. 1–3.
A mixed plasma containing MN/NMN/3-MT at concentrations of 0.038, 0.14, and 0.01 nmol/L, respectively, was diluted and extracted via MBE, and analyzed using LC-MS/MS. The LOQ was defined as the signal-to-noise ratio (S/N) ssion is preCV Vgna% was obtained for more than ten injections; the LOD was defined as S/N ≥ 3. The LOQ for the MBE method is 0.0125 nmol/L, 0.0442 nmol/L, and 0.0103 nmol/L, for MN, NMN, and 3-MT, respectively, with a CV of 9.57%, 13.89%, and 11.57%, respectively; the LOD is 0.0097 nmol/L, 0.0331 nmol/L, and 0.0025 nmol/L for MN, NMN, and 3-MT, respectively. Considering that the reported upper limit for diagnosing PPGLs is in the range 0.13–0.85 nmol/L for MN [6], 0.50–1.39 nmol/L for NMN [6], and 0.03–0.13 nmol/L for 3-MT [13], the current LOQ can meet the requirement for clinical practice.
Precision
The precision was evaluated by measuring two pools of plasma five times each per day on five different days according to CLSI EP-15A2 [14]. The results indicated that the MBE method has good reproducibility with within-run CVs of 1.96–2.00%, 4.06–5.75%, and 3.89–4.90% for MN, NMN, and 3-MT, respectively; whereas the total CVs for MN, NMN, and 3-MT were 1.96–2.80%, 5.12–5.75%, and 5.44–6.27%, respectively (Table 1). According to the allowable limits in the external quality assessment program organized in 2021 by the National Center of Clinical Laboratories in China, the allowable limits for all three analytes are 30%, and our method meets the requirement of 1/2 allowable limits.
Absolute recovery
Absolute recovery is a key parameter that influences the sensitivity; it determines how successfully the method can isolate and collect the compounds of interest [15]. Six mixed patient samples were prepared and each was divided into five aliquots, one was extracted using the MBE as the blank sample, two were spiked with 20 μL of the mixed standards with different concentrations of (MN: 4.53/9.05 nmol/L; NMN: 4.77/9.54 nmol/L; 3-MT: 5.14/10.28 nmol/L) after MBE extraction, and two were spiked with the above-mixed standards before the MBE extraction. The extract was diluted with water to ensure that the final volumes were equal in the final step. The absolute recovery (AR) was calculated using the equation: AR% = (Analyte ResponseBefore MBE extraction−Analyte ResponseBlank)/(Analyte Responseafter MBE extraction−Analyte ResponseBlank)*100. The AR% for MN, NMN, and 3-MT ranged from 41.9 to 53.0%, 23.1 to 46.3%, and 39.2 to 54.9%, respectively. Although the AR was somewhat low, the relative recovery in the following step was approximately 100%. Although the relatively low AR can influence the sensitivity of the method, when the LOQ and the range of the target compound concentrations in the plasma are considered, the sensitivity can meet the clinical requirements.
Relative recovery and accuracy
In the isotope dilution LC-MS/MS method, the AR of some analytes may be low; however, if the IS can correct any losses experienced during the extraction process with the high sensitivity of the LC-MS/MS, the signal is sufficient for the quantification. Herein, we evaluated the relative recovery by spiking known concentrations of metanephrines into two plasma samples (the three theoretical concentrations added for MN, NMN, and 3-MT are listed in Table 2). The samples were extracted via MBE, and analyzed and quantified using LC-MS/MS. The accuracy and relative recovery were evaluated by comparing the added theoretical concentrations with the measured concentration according to our previous formula. The RE% of MN varied from 103.7 to 107.4%, whereas that of NMN varied from 93.5 to 98.9%, and that of 3-MT varied from 95.7 to 100.6%. The bias of the calculated values compared with that of the added theoretical concentrations were all within 10% (Table 2).
Matrix effect
The electrospray ionization (ESI) monitoring mode is sensitive; however, it is also subject to matrix effects, particularly ion suppression. The extraction step is most affected by the matrix effect; however, with an appropriate IS, the matrix effects of ion suppression or enhancement can often be compensated for [15]. In this study, the matrix effect was evaluated by a post-extraction analyte spiking method [15, 16]. Plasma samples from seven individuals were collected, and then each was divided into two aliquots. After extracting the plasma samples using the MBE method, mixed standards (100 μL) of different concentrations were added to each aliquot. The two concentrations of MN, NMN, and 3-MT were 0.45/1.81, 0.48/1.91, and 0.51/2.06 nmol/L. IS (80 μL) was added to each sample. The absolute response (peak area) of the standards in the solvent was compared with that in the MBE-treated plasma. In addition, the relative response ratio of the analyte peak area/internal standard in the solvent was compared with that in the MBE-treated plasma to evaluate whether isotope-labeled IS can compensate for the matrix effect, if any existed. The absolute responses of the standards, without IS correction, in the plasma samples obtained by MBE compared with those in the solvent were 73.0–104.9% (MN), 71.7–106.6% (NMN), and 80.5–104.7% (3-MT) at the level of 0.45/0.48/0.51 nmol/L, and 83.2–104.9% (MN), 68.6–104.5% (NMN), and 88.7–103.8% (3-MT) at the level of 1.81/1.91/2.06 nmol/L. After IS correction, the ratios were 99.1–101.0% (MN), 100.8–103.3% (NMN), and 99.5–100.6% (3-MT) at the level of 0.45/0.48/0.51 nmol/L, and 101.0–102.1% (MN), 101.1–104.6% (NMN), and 100.3–101.0% (3-MT) at the level of 1.81/1.91/2.06 nmol/L. The absolute responses of the analytes indicated that in some samples, ion suppression effects existed without IS correction. However, IS correction compensated for the observed matrix effects.
Comparison between MBE and SPE methods
According to CLSI document EP09 [17], a minimum of 40 samples, preferably patient samples, are recommended for method comparison, which includes different concentrations of the target analytes. In this study, we collected the surplus of 130 plasma samples from clinical requests for MNs measurement. Although the three analytes extracted using SPE exhibited a slope value > 1 compared with that of the samples extracted using MBE, the results of the two methods correlate well with correlations r > 0.99 for MN, NMN, and 3-MT. The mean bias% for MN, NMN, and 3-MT were low, −2.9%, −3.2%, and −3.2%, respectively. The bias% obtained for the MBE method compared with that of SPE were lower than 25% (Fig. 3).

Comparison of MNs and 3-MT results extracted using MBE and traditional solid-phase extraction (SPE) methods. a–c Passing-Bablok regression analyses between MBE and SPE for MN, NMN, and 3-MT, respectively. Blue solid line: Passing-Bablok regression line; purple dotted line: 95% CI for the regression line. d–f Red solid line: diagonal line; Bland-Altman plots for assessing agreement between MBE and SPE for MN, NMN, and 3-MT, respectively. Solid deep blue line: mean difference; green dotted line: 95% CI for mean difference; purple dotted line: limits of agreement (from −1.96 to +1.96SD); solid blue line: 95% CI for limits of agreement line; orange dotted line: zero line
Limitations of the current MBE method
In this first generation of the automated MBE instrument, only 16 samples can be extracted in a 96-deep well plate, and 48 samples (three 96-deep well plates) can be processed simultaneously by using the extraction machine. The throughput is not high. The preliminary steps of adding samples and IS into the pores of the 96-well plate are either manual or require other liquid transferring instruments. However, the principle of the MBE enables the automated use of LC-MS/MS similar to the biochemical or immunoassay analyzer instruments. The next generation of instruments based on the MBE principle is in progress and should make sample preparation more convenient and with higher throughput. We predict that adding samples and IS will also be integrated into the system.
Discussion
MNs are the initial biochemical markers for diagnosing PPGL, and LC-MS/MS is recommended as the first choice for measuring MNs [6]. Although 3-MT is reported to be associated with the metastatic characteristic of PPGLs [13, 18], there are no mature immunoassay or chemical methods for measuring MNs and 3-MT. The newly developed MBE method for MNs and 3-MT exhibited excellent performance with symmetrical chromatography peaks, good linearity, precision, comparability, etc. In addition, the MBE method is comparable with the traditional SPE method with a correlation > 0.99.
Several approaches have been developed to simplify the sample pretreatment of MNs and other analytes prior to using LC-MS/MS. For example, some laboratories use liquid handling systems based on LLE or SPE, etc., and others use online or offline SPE [8, 19]. These efforts advanced the use of LC-MS/MS in clinical laboratories; however, a few limitations remained, including a need for highly trained experts, complicated steps, centrifugation, and positive pressure instruments. The current automation of LC-MS/MS is underdeveloped compared with that of standard clinical chemistry or immunoassay analyzers. A fully automated LC-MS/MS system similar to the current clinical chemistry or immunoassay analyzers would promote the analytical potential of LC-MS/MS to clinical laboratory staff.
LC-MS/MS has been recently used more often in clinical practice; some guidelines even recommend LC-MS/MS as their initial choice because of its high specificity. Although the LC-MS/MS method has a number of advantages, such as high specificity, good sensitivity, and multiple compound quantification, the traditional LLE, SPE sample preparation before LC-MS/MS analysis needs highly skilled operators, is time-consuming, and contains many manual handling protocols [8]. These disadvantages have limited the use of LC-MS/MS in clinical practice. Laboratory staff members require simple and automated instruments for the pretreatment of samples. For LC-MS/MS to become a routine part of clinical chemistry operations, the in vitro diagnostic medical device (IVD) industry companies must strive to automate the sample preparation and develop a “push-button-design” for the future [20]. Recently, there has been a development to this end; some laboratories use automatic smart liquid transferring workstations such as Tecan Freedom EVO and Hamilton Microlab STAR to resolve the challenges of manual handling; however, the current liquid transferring working stations are usually based on traditional LLE or SPE and are often complicated and not very flexible. The newly developed MBE overcame the liquid transferring process during the traditional LLE or SPE. Instead of transferring the target in the liquid, it transfers magnetic beads using a magnetic bar, the magnetic bead surface is modified using chemical bonds, based on the target analytes, and the targets are adsorbed and transferred on the magnetic beads by a magnetic bar. The magnetic beads used in the MBE method are different from the material used in the immunoassay method where antibodies are combined; and the principle of transferring magnetic beads in different solvents without positive pressure instrumentation or centrifugation proves that with automation MBE could be a valuable method similar to the immunoassay or chemical analyzers.
The measurement uncertainties of the new extraction method based on magnetic beads were evaluated according to the ISO Guide Uncertainty of measurement. For two concentrations of plasma samples, the estimated relative expanded uncertainties (k= 2) for MN, NMN, and 3-MT were 8.91–8.93%, 8.13–8.55%, and 5.91–6.04%, respectively (Supplemental Table 1). The potential sources of uncertainty arose from the variation of repeated measurement, purity of the standard material, preparation of the standard solutions, sampling of the sample and IS, and possible system errors (using the biggest bias from the data of relative recovery showed in Table 2). The data contained within Supplemental Table 1 indicates that possible system errors were the largest source of uncertainty, accounting for more than 80% of the total uncertainty. The other factors contributing to the uncertainty were errors incurred in the sampling and repeated measurements. If the possible system errors were excluded, the uncertainties of the MN, NMN, and 3-MT methods were 2.52–2.60%, 3.12–4.09%, and 3.20–3.43%, respectively.
Conclusions
In this study, a new extraction method based on magnetic beads was developed and validated. We found that the performance of the newly developed method can meet clinical requirements by validating the extraction of MNs and 3-MT using the MBE method; the performance of the MBE method was comparable with that of the traditional SPE method. The measurement of MNs and 3-MT has been previously accomplished; however, the principle based on magnetic beads mimicking SPE is novel. It does not require centrifugation and positive pressure, and it has the potential to enable the automation of sample preparation for LC-MS/MS. Although there are limitations to the current extraction instrument, the MBE principle enables LC-MS/MS to be automated in a manner similar to biochemical or immunoassay methods. This study may mark a new era for liquid chromatography tandem mass spectrometry in clinical application.
References
Shi J, Dhaliwal P, Zheng Y Zi, Straseski JA, Cervinski MA, Shajani-Yi Z, et al. An intact ACTH LC-MS/MS assay as an arbiter of clinically discordant immunoassay results. Clin Chem. 2019; 10.1373/clinchem.2019.306365
Kane J, Middle J, Cawood M. Measurement of serum testosterone in women; what should we do? Ann Clin Biochem. 2007. https://doi.org/10.1258/000456307779595896.
Wright MJ, Thomas RL, Stanford PE, Horvath AR. Multiple reaction monitoring with multistage fragmentation (MRM3) detection enhances selectivity for LC-MS/MS analysis of plasma free metanephrines. Clin Chem. 2015. https://doi.org/10.1373/clinchem.2014.233551.
Lagerstedt SA, O'Kane DJ, Singh RJ. Measurement of plasma free metanephrine and normetanephrine by liquid chromatography-tandem mass spectrometry for diagnosis of pheochromocytoma. Clin Chem. 2004;https:// doi:https://doi.org/10.1373/clinchem.2003.024703
Virag D, Kiraly M, Drahos L, Edes AE, Gecse K, Bagdy G, et al. Development, validation and application of LC-MS/MS method for quantification of amino acids, kynurenine and serotonin in human plasma. J Pharm Biomed Anal. 2020. https://doi.org/10.1016/j.jpba.2019.113018.
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014. https://doi.org/10.1210/jc.2014-1498.
Stanczyk FZ, Clarke NJ. Advantages and challenges of mass spectrometry assays for steroid hormones. J Steroid Biochem Mol Biol. 2010. https://doi.org/10.1016/j.jsbmb.2010.05.001.
Vogeser M, Kirchhoff F. Progress in automation of LC-MS in laboratory medicine. Clin Biochem. 2011. https://doi.org/10.1016/j.clinbiochem.2010.06.005.
van Faassen M, Bischoff R, Eijkelenkamp K, de Jong WHA, van der Ley CP, Kema IP. In matrix derivatization combined with LC-MS/MS results in ultrasensitive quantification of plasma free metanephrines and catecholamines. Anal Chem. 2020. doi:https://doi.org/10.1021/acs.analchem.0c01263
Yu S, Yin Y, Li Q, Yu J, Liu W, Wang D, et al. Validation of an improved liquid chromatography tandem mass spectrometry method for rapid and simultaneous analysis of plasma catecholamine and their metabolites. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1129: 121805. https://doi.org/10.1016/j.jchromb.2019.121805.
Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010. https://doi.org/10.1097/MPA.0b013e3181ebb4f0
Wayne P (2014) Liquid chromatography-mass spectrometry methods; Approved Guidelines. CLSI. Clinical and Laboratory Standards Institute;CLSI document C62-A.
Peitzsch M, Prejbisz A, Kroiss M, Beuschlein F, Arlt W, Januszewicz A, et al. (2013) Analysis of plasma 3-methoxytyramine, normetanephrine, and metanephrine by ultraperformance liquid chromatography-tandem mass spectrometry: utility for diagnosis of dopamine-producing metastatic phaeochromocytoma. Ann Clin Biochem. https://doi.org/10.1258/acb.2012.012112
Wayne,CLSI (2005) CLSI document EP15-A2. User verification of performance for precision and trueness ag, 23d, CLSI
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. https://doi.org/10.1021/ac020361s
Yadav M, Trivedi V, Upadhyay V, Shah G, Baxi GA, Goswami S, et al. Comparison of extraction procedures for assessment of matrix effect for selective and reliable determination of atazanavir in human plasma by LC-ESI-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. https://doi.org/10.1016/j.jchromb.2011.12.031
NCCLS (2002) Method comparison and bias estimation using patient samples; Approved Guideline—Second Edition. NCCLS document EP9-A2
Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012. https://doi.org/10.1016/j.ejca.2011.07.016.
de Jong WH, Graham KS, van der Molen JC, Links TP, Morris MR, Ross HA, et al. (2007) Plasma free metanephrine measurement using automated online solid-phase extraction HPLC tandem mass spectrometry. Clin Chem. https://doi.org/10.1373/clinchem.2007.087114
Seger C, Salzmann L. After another decade: LC-MS/MS became routine in clinical diagnostics. Clin Biochem. 2020. https://doi.org/10.1016/j.clinbiochem.2020.03.004.
Funding
This work was funded by the National Key Research and Development Program of China (No. 2021YFC2401100); Innovation-Led Industrial Cluster Project of Zheng-Luo-Xin National Innovation Demonstration Area (201200211100); and Beijing Natural Science Foundation (No.7212087).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design, material preparation, and data collection and analysis. The first draft of the manuscript was written by Songlin Yu and all authors commented on previous versions of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Ethics Committee of Peking Union Medical College & Chinese Academy of Medical Sciences, Peking Union Medical College Hospital (JS-2956)
Conflict of interest
The authors declare no competing interests.
Source of biological material
Plasma for the method validation and comparison was obtained from that remaining from patient samples in the Department of Laboratory Medicine, Peking Union Medical College Hospital.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, S., Zhou, W., Yu, J. et al. An automated magnetic bead extraction method for measuring plasma metanephrines and 3-methoxytyramine using liquid chromatography tandem mass spectrometry. Anal Bioanal Chem 414, 3541–3549 (2022). https://doi.org/10.1007/s00216-022-03984-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-03984-x