
Abstract
In this study, a simple and rapid sample preparation method named liquid-nitrogen-induced homogeneous liquid–liquid microextraction has been developed for the extraction and pre-concentration of Co(II) and Ni(II) ions before their analysis by flame atomic absorption spectrometry. For this purpose, first, acetonitrile containing 8-hydroxyquinoline is added into a sample solution and the mixture is vortexed. As a result, a homogeneous solution is formed. Subsequently, the solution is cooled using liquid nitrogen for a few seconds. By this process, due to difference in the freezing point of acetonitrile and water, the homogeneous state is broken and the analytes (as oxinate complexes) are extracted into liquid acetonitrile phase collected on top of the frozen aqueous phase. The linear dynamic ranges obtained for Ni(II) and Co(II) were 1.0–30 and 0.50–20 μg L−1, respectively. The obtained limits of detection were 0.36 and 0.20 μg L−1 for Ni(II) and Co(II), respectively.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, environmental contamination resulting from heavy metal ions has become a food safety concern worldwide, because of their persistence, carcinogenic effects, high toxicity even at low concentrations, and global distribution in soil, water, and air. Also, the ions tend to accumulate in food chains with a low decomposition rate [1,2,3]. The main sources of these elements in the environment are industrial, pharmaceutical, domestic, and agricultural effluents [4]. Heavy metals can affect central nervous system function and mental development, change the blood composition, and disturb the operation of some human body organs like the heart, lungs, liver, and kidneys [5]. As a result, the investigation and determination of heavy metals at trace levels in the food chain and environmental samples is one of the targets of analytical chemists, due to the important roles of heavy metals in our lives [6]. Cobalt has both beneficial and harmful effects on human health. Vitamin B12, a tetrapyrrole complex containing a cobalt ion, plays an important role in metabolic processes and protein synthesis [7, 8]. It is a naturally occurring element found in water and plants, and is also used to produce alloys used in industry. But it may bring about several health problems such as diarrhea, paralysis, lung irritation, low blood pressure, and bone defects [9, 10]. The toxic effects of nickel are well known and it is highly irritant to the skin, and is toxic to the cardiovascular system as well as being carcinogenic [11, 12]. Therefore, accurate and precise determination of these metallic ions in environmental samples such as water and fruit juice samples is a major challenge for analytical chemists.
Numerous instrumental techniques such as inductively coupled plasma-mass spectrometry [13], flame atomic absorption spectrometry (FAAS) [14, 15], graphite furnace atomic absorption spectrometry [16, 17], atomic fluorescence spectrometry [18, 19], and induced coupled plasma-optical emission spectrometry [20] have been used for the sensitive and selective detection of metal ions. Although all of these instrumental techniques have been useful in tracing metal ions, direct determination of heavy metal ions in real samples is not feasible because of their low level of concentration and matrix complexity of samples [21].
To overcome these limitations, the extraction and pre-concentration of heavy metal ions are usually required prior to their measurement by analytical instruments [22]. During the past few decades, various methods have been established to extract and pre-concentrate trace amounts of heavy metals such as liquid–liquid extraction [23], solid-phase extraction [24, 25], coprecipitation [26], and cloud point extraction [27,28,29]. All of these methods have drawbacks such as time-consuming and tedious processes, and the consumption of large amounts of toxic and expensive organic solvents. Thus, recent trends in analytical chemistry have been towards the simplification and miniaturization of sample preparation procedures in order to reduce the amount of organic solvents and replace chlorinated solvents with those that are more environmentally friendly [30]. Homogeneous liquid–liquid microextraction (HLLME) is a powerful method which has attracted a great deal of attention owing to its simplicity of operation, rapidity, and low cost for the extraction and pre-concentration of various analytes. In the present method, a water-miscible extraction solvent, mostly acetonitrile (ACN), is mixed with an aqueous sample solution containing the analytes to form a homogeneous solution which is subsequently broken by adding a phase separation agent [31]. In the ACN/water-based HLLME, the homogeneous solution can be separated into two phases by the addition of phase separation agents such as sugars [32] and salts [33], or cooling the solution [34]. In the case of the last mode, at low temperatures (< −20 °C), the solubility of ACN in the aqueous phase decreases, and ACN phase containing the analytes is separated from the aqueous solution as the upper layer. The main advantage of this method is that, unlike the other modes, it does not require any foreign inducer substance to break the homogeneous state, which can reduce the cost and avoid the entrance of new impurities. Up to now, this method has been used for the cleanup, extraction, and pre-concentration of the target analytes in samples such as milk [35], sticky traditional Chinese patent medicine [36], meat [37], and tea [38]. The major problem with the methods mentioned above is the time-consuming cooling procedure (requires between 1 and 12 h).
The aim of this study is to develop a sample preparation method based on liquid-nitrogen-induced homogeneous liquid–liquid microextraction for the extraction and pre-concentration of Co(II) and Ni(II) ions prior to their determination with FAAS. In this work, 8-hydroxyquinoline (8-HQ) is used as a chelating agent. In the proposed method, there is no need to dilute samples in order to reduce the matrix effect, which can enhance detection limits of the method. In addition, a prompt phase separation occurs in a few seconds due to use of liquid nitrogen. No toxic organic solvents such as chlorinated or aromatic solvents are used in the extraction procedure. Ease of operation, being environmentally friendly, rapidity, and low cost can be considered the main advantages of the introduced approach.
Materials and methods
Chemicals and solutions
A stock mixture solution of Co(II) and Ni(II) containing 100 mg L−1 of each cation was prepared by dissolving suitable amounts of Co(NO3)2·6H2O and Ni(NO3)2·6H2O (Merck, Darmstadt, Germany) in deionized water (Ghazi Company, Tabriz, Iran). A working standard solution (5 μg L−1 of each) was prepared daily by diluting the stock solution with deionized water. Also, a mixture standard solution with a concentration of 1 mg L−1 of each analyte in deionized water was prepared and injected into the FAAS each day (three times) for quality control, and the obtained signals were used to calculate enrichment factors (EFs) and extraction recoveries (ERs) of the analytes. ACN, 8-HQ, sodium chloride, hydrochloric acid (37%, w/w), nitric acid, and sodium hydroxide with highest purity were supplied from Merck.
Real samples
Mineral water, fruit juice, and soda samples were supplied from local supermarkets (Tabriz, Iran). They were used without pretreatment. The accuracy of the presented procedure was checked by the analysis of a certified reference material (CRM); SPS-WW2 waste water, batch no. 108, Spectrapure Standards AS (Oslo, Norway). Because of high concentrations of Co(II) and Ni(II) in the CRM, it was diluted 100 and 200 times, respectively.
Instrumentation
A Shimadzu AA-6300 (Kyoto, Japan) FAAS was used for absorbance measurements. It possessed a 100-mm burner head and deuterium background correction. The radiation sources used were cobalt and nickel hollow cathode lamps which operated at currents of 20 and 25 mA, and wavelengths of 240.7 and 232.0 nm, respectively. The flame was generated with a mixture of air and acetylene with flow rates of 15 and 2.3 L min−1, respectively. Quantitative analysis of cobalt(II) and nickel(II) in some samples was also performed with a Shimadzu 6300 atomic absorption spectrometer (Kyoto, Japan) equipped with a heated graphite tube atomizer. A vortex mixer (Labinco L46, Netherlands) was used for shaking during the extraction process. pH measurements were performed with a Metrohm pH meter model 654 (Herisau, Switzerland).
Procedure
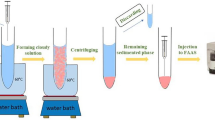
Five milliliters of the mixed standard solution of the analytes (5 μg L−1) or real sample was transferred into a 10-mL glass test tube. Then 1.2 mL ACN containing 8-HQ (0.1 M) as a complexing agent was added into the solution. The mixture was vortexed for 30 s, and a homogeneous solution was formed. The tube was then placed into liquid nitrogen for 10 s. Due to the difference between the freezing point of ACN and water, the homogeneous state was broken, and the aqueous phase was frozen at the bottom of the tube, whereas ACN (200 ± 5 μL) containing the complexed cations was collected as a liquid phase on top of the frozen phase. Subsequently, two 90-μL portions of the collected ACN phase were removed and injected into the FAAS separately using a homemade micro-sample introduction system [39].
Graphite furnace atomic absorption spectrometry (GFAAS) method
To evaluate the accuracy of the developed method, concentrations of Co(II) and Ni(II) in some real samples were determined by another method [41, 41]. For this purpose, 10 mL of each sample was placed into a high-form porcelain crucible and heated at 90 °C. After evaporation of the solvents, the remaining residues were heated in a furnace at 450 °C for 8 h. The ashed samples were dissolved in HNO3 (65%, v/v) and filtered through Whatman filter paper. The filtrate was collected in a 10-mL volumetric flask and made up to volume with deionized water. This clear solution was injected into the GFAAS to determine the concentration of the analytes.
Calculation of EF and ER
EF is defined as the ratio between the analyte concentration in the collected ACN phase (Ccol) and the initial concentration of analyte (C0) in the sample:
where Ccol is obtained from a calibration graph. The percentage of the total analyte amount (n0) that is transferred into the collected ACN phase (ncol) is defined as ER.
In this formula, Vaq and Vcol represent the volumes of the aqueous solution and the collected ACN phase, respectively.
Results and discussion
Study of type and volume of extraction solvent
In HLLME procedures, the type and volume of the extraction solvent are essential factors for an effective extraction. In this study, the solvent should possess some merits such as capability for extraction of the formed complexes, miscibility with aqueous phase to form a homogenous solution, and ability to form a two-phase system at low temperature. According to these properties and previous reports [34, 37], three common solvents used in HLLME, namely acetone, methanol, and ACN, were evaluated. The experiments showed that only ACN forms a two-phase system with an aqueous solution upon lowering the temperature. It is well known that the nitrile group of ACN exhibits proton acceptor properties. On the other hand, its methyl group is probably a proton donor [42]. Therefore the molecules of ACN probably form dimers in the solution with decreasing temperature, and ACN-water hydrogen bonds are progressively replaced by dipole-dipole interactions between polar C≡N groups. Therefore, in this study, ACN was used as an extraction solvent. The effect of ACN volume on the extraction efficiency of the method was investigated in the range of 0.4–2.0 mL. Based on the obtained results, at the volume of 0.4 mL ACN, no phase was separated by cooling the solution, and the method became useless. On the other hand, in the case of coupling such microextraction system with FAAS, it should be taken into account that the required injected volume into the burner/nebulization system is 90 μL for each cation for effective atomization and quantification. The experiments showed that in the amounts lower than 1.2 mL, the collected ACN phase volume was less than 180 μL, and a systematic error was generated. Also, the highest extraction efficiency of the studied ions was observed for 1.2 mL of ACN (Fig. S1 in Electronic Supplementary Material, ESM). However, the decrease in the extraction efficiency at higher volumes of ACN can be attributed to the dilution effect. Hence, 1.2 mL of ACN was used as the optimal extraction solvent volume for pre-concentration of the ions for further experiments.
Optimization of 8-HQ concentration
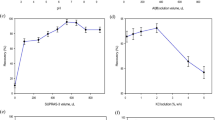
In the present work, the type and concentration of the appropriate chelating agent is very important, because it should be able to dissolve in ACN and react with the selected metallic ions to form stable complexes. For this purpose, 8-HQ was used as a chelating agent, and its concentration was investigated ranging from 0.01 to 0.50 mol L−1. The results (Fig. 1) showed that the ERs of the selected ions were enhanced up to 0.10 mol L−1, and at higher concentrations, they were approximately constant. Therefore, 0.10 mol L−1 of 8-HQ was used as the optimal concentration of the chelating agent for further experiments.

Effect of 8-HQ concentration on the extraction recovery of the analytes. Extraction conditions: sample, 5 mL deionized water spiked with 5 μg L−1 each of Co(II) and Ni(II); extraction solvent (volume): ACN (1.2 mL); and vortexing time: 20 s. The error bars represent standard deviations (n = 3)
Effect of pH
The efficiency of the proposed method in the extraction of the studied cations can be influenced by varying the pH of the aqueous phase, due to the effect of pH on the formation of the cation complexes as well as the chemical forms of Co(II) and Ni(II) ions. Therefore, the effect of sample pH on the ER of the selected heavy metals was investigated in the pH range of 2–12 by adding appropriate amounts of 1 mol L−1 hydrochloric acid or sodium hydroxide solution. The results in Fig. 2 show that the ERs increased with the enhancement of pH from 2 to 4, and then nearly remain constant up to 8. Decreasing ERs at high pH values are probably due to the hydrolysis of the cations in highly basic solutions. As a result, the efficiency of the method was pH-independent in the pH range of 4–8. It should be noted that the pH of all samples used in this study was between 4 and 8; therefore, the original samples were used without pH adjustment.

Effect of sample pH on the extraction recovery of the analytes. Extraction conditions are the same as those used in Fig. 1, except the concentration of 8-HQ was selected as 0.10 M. The error bars represent standard deviations (n = 3)
Effect of ionic strength
From the theoretical perspective, addition of a salt can affect the extraction efficiency of the analytes through two different ways: (1) salting-out effect; addition of a salt decreases the analyte's solubility in the aqueous phase through increasing polarity of the aqueous solution and finally improves the extraction efficiency of various extraction methods; and (2) salting-in effect; addition of a salt can increase the viscosity of the aqueous solution which leads to the decreased diffusion coefficients and ERs of the analytes. Accordingly, the effect of this parameter on the extraction efficiency was investigated by using different concentrations of sodium chloride in the aqueous phase in the range of 0–15% (w/v). In this work, at concentrations higher than 10% (w/v), the freezing point of the aqueous solution decreased near the freezing point of ACN. Therefore, the phase separation due to the difference in freezing temperature was difficult. The results in Fig. S2 (in ESM) indicate that ERs of the analytes are constant up to 2% (w/v) NaCl. However, by increasing the concentration of NaCl from 2 to 10% (w/v), the analytical signals decreased due to the increase in the volume of ACN collected and dilution effect. Therefore, the subsequent studies were performed without salt addition.
Optimization of vortexing time
Vortex mixing plays an effective role in shortening the extraction time and also quantitative pre-concentration of the selected cations by a complete dispersion of 8-HQ into the aqueous phase. To investigate the effect of vortex mixing time on the extraction efficiency, it was tested in the range of 5–50 s. The results (Fig. S3 in ESM) indicate that the mixing of solution in this step has a positive impact on the extraction efficiency. Hence, the vortex mixing time equal to 30 s was chosen for further experiments.
Optimization of cooling time
In this study, for the first time, liquid nitrogen (−196 °C) was selected as a coolant to shorten the extraction time of the studied cations. When the homogenous solution of ACN-water was placed into the liquid nitrogen for a relatively long time, both the aqueous phase and ACN were frozen. In this case, it is necessary to hold the tube containing the frozen phases at room temperature for several minutes to defreeze the ACN phase while the aqueous phase remains in the frozen state. Therefore, to shorten the extraction time, the cooling time (the time of placement of the homogenous solution into the liquid nitrogen) was studied. It was observed that when the cooling time was in the range of 10–20 s, phase separation and freezing the aqueous phase were completed. According to the obtained results, phase separation did not occur at the cooling times lower than 8 s; additionally, freezing of the ACN phase did not occur at the cooling times longer than 25 s. So, 10 s was selected as the cooling time for subsequent studies.
Study of coexisting ions
To assess possible analytical applications of the developed procedure for determination of Co(II) and Ni(II) in the studied samples, the effect of some foreign ions which could coexist in the real samples was studied. In these experiments, the extraction of the analytes was investigated in 5.0 mL of aqueous solutions containing 2 μg L−1 and 10 μg L−1 each of Co(II) and Ni(II) in the presence of different concentrations of the other potentially interfering ions. A certain species was considered to interfere if it resulted in a ± 5% variation in the ERs of the analytes. The results are listed in Table 1. They indicate that the presence of most of the foreign ions had negligible disturbance in the determination of Co(II) and Ni(II) under the optimized conditions.
Analytical features
Under the optimal experimental conditions, the analytical characteristics of the proposed method including limit of determination (LOD), limit of quantification (LOQ), coefficient of determination (r2), linear range (LR), and relative standard deviation (RSD) of the analytes were evaluated and are summarized in Table 2. The LODs were calculated as 3SB/m (SB is the standard deviation of the blank, and m is the slope of the calibration graph), and the obtained amounts for Co(II) and Ni(II) were 0.20 and 0.36 μg L−1, respectively. The repeatability of the method was assessed by analyzing six separate standard solutions (2.5 μg L−1 of each cation), and it was found that RSD values were less than 4% for intra- (n = 6) and inter-day (n = 6). The EFs and ERs for the analytes were about 24 and 96%, respectively. The LOQs (calculated from 10SB/m) were 0.5 and 0.8 μg L−1 for Co(II) and Ni(II), respectively. Also, the r2 values (≥ 0.994) verified the good linearity of the proposed method throughout the studied concentrations. The low LODs and LOQs and the good repeatability show that the present method is efficient in determination of the studied cations.
Analysis of real samples
To assess the efficiency of the developed method in analysis of the analytes in real samples, it was used under the optimal conditions for the analysis of several samples including mineral water, peach juice, orange juice, mango juice, pomegranate juice, and soda. The samples were spiked with three concentrations of the analytes at levels of 2, 5, and 15 μg L−1 (of each) in order to evaluate relative recoveries and the matrix effect. According to the obtained results (Table 3), the matrices of the samples had no significant effect on the performance of the suggested approach. The pomegranate juice was free of the analytes. In other samples, one of the analytes was determined at a μg L−1 level. The obtained concentrations of the analytes in the samples are given in Table 3.
Method validation
To evaluate the accuracy of the developed method, two series of experiments were carried out: (1) analysis of Co(II) and Ni(II) in a CRM; the obtained concentrations were 309.4 ± 7.2 and 4975.2 ± 116.8 μg L−1 for cobalt(II) and nickel(II), respectively, which were in good agreements with the certified values, 300 ± 2 and 5000 ± 25 μg L−1 for cobalt(II) and nickel(II), respectively, and no significant difference (ttest, obtained = 0.7, t0.05,2 = 4.30) was observed, and (2) analysis of the analytes in three real samples including peach juice, orange juice, and mango juice by the other method [see “Materials and methods” section]. The results are shown in Table 4. Comparison of the concentrations obtained with the two methods shows that there is no significant difference between the results.
Comparison of the developed method with other approaches
A comparison between the proposed method and some recently published works for pre-concentration and determination of Co(II) and Ni(II) ions in various samples is shown in Table 5. Most of the analytical methods mentioned in the Table 5 are time-consuming, while the proposed procedure is fast and low-cost. As it can be seen, the LR, LOD, and RSD values of the proposed method are better than or comparable to those reported for the other methods. All these results illustrate that the proposed method can be used as a sensitive and simple extraction method, and is capable of generating accurate and precise results for analysis of ultra-trace amounts of Co(II) and Ni(II) in aqueous samples.
Conclusions
This paper describes the development of an HLLME technique based on liquid nitrogen-induced phase separation for the extraction and pre-concentration of Co(II) and Ni(II) ions in fruit juice and water samples, followed by FAAS determination. In this work, ACN was used as an extraction solvent, which allowed for minimal use of organic solvents, in turn reducing the risk of adverse effects on human health and the environment. In addition, the proposed method was fast because of the use of liquid nitrogen in phase separation, which reduced the extraction time. The obtained experimental results indicated that the developed procedure provided good precision and accuracy, low LODs, and high ERs. These results revealed that the suggested approach can be applied as a low-cost and rapid analytical method in toxicological and risk assessment studies.
Abbreviations
- EF:
-
Enrichment factor
- ER:
-
Extraction recovery
- FAAS:
-
Flame atomic absorption spectrometry
- HLLME:
-
Homogeneous liquid–liquid microextraction
- 8-HQ:
-
8-Hydroxyquinoline
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
References
Abbasi S, Roushani M, Khani H, Sahrae R, Mansouri G. Synthesis and application of ion-imprinted polymer nanoparticles for the determination of nickel ions. Spectrochim Acta Part A. 2015;140:534–43.
Abdullah BA, Talpur FN, Kumar A, Shah MT, Mahar AM, Amina. Synthesis of ultrasonic-assisted lead ion imprinted polymer as a selective sorbent for the removal of Pb2+ in a real water sample. Microchem J. 2019;146:1160–8.
Branger C, Meouche W, Margaillan A. Recent advances on ion-imprinted polymers. React Funct Polym. 2013;73:859–75.
Werner J. Ionic liquid ultrasound-assisted dispersive liquid-liquid microextraction based on solidification of the aqueous phase for preconcentration of heavy metals ions prior to determination by LC-UV. Talanta. 2018;182:69–73.
Fiamegos Y, Vahcic M, Emteborg H, Snell J, Raber G, Cordeiro F, et al. Determination of toxic trace elements in canned vegetables. The importance of sample preparation. Trends Anal Chem. 2016;85:57–66.
Buhani, Narsito, Nuryono, Kunarti ES. Production of metal ion imprinted polymer from mercapto-silica through sol-gel process as selective adsorbent of cadmium. Desalination. 2010;251:83–9.
Ford SH, Nichols A, Gallery JM. Separation and study of corrinoid cobalt-ligand isomers by high-performance liquid chromatography. J Chromatogr A. 1991;536:185–91.
Bernard MA, Nakonezny PA, Kashner TM. The effect of vitamin B12 deficiency on older veterans and its relationship to health. J Am Geriatr Soc. 1998;46:1199–206.
Bratter P, Schramel P. Trace elements in analytical chemistry, medicine and biology. New York: Walter de Gruyter; 1980.
Tsalev DL, Zaprianov ZK. Atomic absorption in occupational and environmental health practice, analytical aspects and health significance. Boca Raton: CRC Press; 1983. vol. 11.
Okumuş V, Özdemir S, Kılınç E, Dündar A, Yüksel U, Baysal Z. Preconcentration with bacillus subtilis-immobilized amberlite XAD-16: determination of Cu2+ and Ni2+ in river, soil, and vegetable samples. Bioremediat J. 2015;19:47–55.
Babos DV, Barros AI, Ferreira EC, Neto JAG. Evaluation of solid sampling for determination of Mo, Ni, Co, and V in soil by high-resolution continuum source graphite furnace atomic absorption spectrometry. Spectrochim Acta B. 2017;130:39–44.
Shen M, Chen L, Han W, Ma A. Methods for the determination of heavy metals in indocalamus leaves after different preservation treatment using inductively coupled plasma mass spectrometry. Microchem J. 2018;139:295–300.
Anthemidis AN, Ioannou KIG. Sequential injection ionic liquid dispersive liquid–liquid microextraction for thallium preconcentration and determination with flame atomic absorption spectrometry. Anal Bioanal Chem. 2012;404:685–91.
Arpa Ç, Arıdaşır I. Ultrasound assisted ion pair based surfactant-enhanced liquid–liquid microextraction with solidification of floating organic drop combined with flame atomic absorption spectrometry for preconcentration and determination of nickel and cobalt ions in vegetable and herb samples. Food Chem. 2019;284:16–22.
Sorouraddin SM, Farajzadeh MA, Okhravi T. Cyclohexylamine as extraction solvent and chelating agent in extraction and preconcentration of some heavy metals in aqueous samples based on heat induced homogeneous liquid-liquid extraction. Talanta. 2017;175:359–65.
Tokman N, Akman S, Bakircioglu Y. Preconcentration of nickel and cobalt prior to their determination by graphite furnace atomic absorption spectrometry using the water-soluble polymer poly(vinyl pyrrolidinone). Microchim Acta. 2004;146:31–4.
Felix CSA, Silva DG, Andrade HMC, Riatto VB, Victor MM, Ferreira SLC. An on-line system using ion-imprinted polymer for preconcentration and determination of bismuth in seawater employing atomic fluorescence spectrometry. Talanta. 2018;184:87–92.
Zeng C, Jia Y, Lee Y, Hou X, Wu L. Ultrasensitive determination of cobalt and nickel by atomic fluorescence spectrometry using APDC enhanced chemical vapor generation. Microchem J. 2012;104:33–7.
Ozdemir S, Yalcin MS, Kilinc E, Soylak M. Boletus edulis loaded with γ-Fe2O3 nanoparticles as a magnetic sorbent for preconcentration of Co(II) and Sn(II) prior to their determination by ICP-OES. Microchim Acta. 2018;185:73-78.
Ghazaghi M, Mousavi HZ, Rashidi AM, Shirkhanloo H, Rahighi R. Graphene-silica hybrid in efficient preconcentration of heavy metal ions via novel single-step method of moderate centrifugation-assisted dispersive micro solid phase extraction. Talanta. 2016;150:476–84.
Stanisz E, Werner JA, Zgoła-Grześkowiak A. Liquid-phase microextraction techniques based on ionic liquids for preconcentration and determination of metals. Trends in Anal Chem. 2014;61:54–66.
Radhika S, Kumar BN, Kantam ML, Reddy BR. Liquid-liquid extraction and separation possibilities of heavy and light rare-earths from phosphoric acid solutions with acidic organophosphorus reagents. Sep Purif Technol. 2010;75:295–302.
Yamini Y, Safari M. Modified magnetic nanoparticles with catechol as a selective sorbent for magnetic solid phase extraction of ultra-trace amounts of heavy metals in water and fruit samples followed by flow injection ICP-OES. Microchem J. 2018;143:503–11.
Zhou Q, Xing A, Zhao K. Simultaneous determination of nickel, cobalt and mercury ions in water samples by solid phase extraction using multiwalled carbon nanotubes as adsorbent after chelating with sodium diethyldithiocarbamate prior to high performance liquid chromatography. J Chromatogr A. 2014;1360:76–81.
Balladares E, Jerez O, Parada F, Baltierra L, Hernández C, Araneda E, et al. Neutralization and co-precipitation of heavy metals by lime addition to effluent from acid plant in a copper smelter. Miner Eng. 2018;122:122–9.
López-García I, Marín-Hernández JJ, Hernández-Córdoba M. Graphite furnace atomic absorption spectrometric determination of vanadium after cloud point extraction in the presence of graphene oxide. Spectrochim Acta Part B. 2018;143:42–7.
Bosch Ojeda C, Sánchez RF. Separation and preconcentration by a cloud point extraction procedure for determination of metals: an overview. Anal Bioanal Chem. 2009;394:759–82.
Manzoori JL, Bavili-Tabrizi A. Cloud point preconcentration and flame atomic absorption spectrometric determination of cobalt and nickel in water samples. Microchim Acta. 2003;141:201–7.
Sadeghi M, Nematifar Z, Irandoust M, Fattahi N, Hamzei P, Barati A, et al. Efficient and selective extraction and determination of ultra trace amounts of Hg2+ using solid phase extraction combined with ion pair based surfactant-assisted dispersive liquid–liquid microextraction. RSC Adv. 2015;5:100511–21.
Anthemidis AN, Ioannou KG. Recent developments in homogeneous and dispersive liquid-liquid extraction for inorganic elements determination: A review. Talanta. 2009;80:413–21.
Shishov A, Nechaeva D, Moskvin L, Andruch V, Bulatov A. Automated solid sample dissolution coupled with sugaring-out homogenous liquid-liquid extraction. Application for the analysis of throat lozenge samples. J Mol Liq. 2017;233:149–55.
Mohebbi A, Farajzadeh MA, Yaripour S, Afshar Mogaddam MR. Determination of tricyclic antidepressants in human urine samples by the three–step sample pretreatment followed by HPLC-UV analysis: an efficient analytical method for further pharmacokinetic and forensic studies. EXCLI J. 2018;17:952–63.
Yoshida M, Akane A. Subzero–temperature liquid-liquid extraction of benzodiazepines for high performance liquid chromatography. Anal Chem. 1999;71:1918–21.
Goulart SM, de Queiroz MELR, Neves AA, de Queiroz JH. Low-temperature clean-up method for the determination of pyrethroids in milk using gas chromatography with electron capture detection. Talanta. 2008;75:1320–3.
Zhang H, Li S, Liu X, Yuan F, Liang Y, Shi Z. Determination of five anthraquinone derivatives in sticky traditional Chinese patent medicines by subzero–temperature liquid-liquid extraction combined with high–performance liquid chromatography. J Liq Chromatogr Relat Technol. 2015;38:584–90.
Ahmadi F, Shahbazi Y, Karami N. Determination of tetracyclines in meat using two phases freezing extraction method and HPLC–DAD. Food Anal Methods. 2015;8:1883–91.
Wang F, Li S, Feng H, Yang Y, Xiao B, Chen D. An enhanced sensitivity and cleanup strategy for the nontargeted screening and targeted determination of pesticides in tea using modified dispersive solid-phase extraction and cold-induced acetonitrile aqueous two-phase systems coupled with liquid chromatography–high resolution mass spectrometry. Food Chem. 2019;275:530–8.
Sorouraddin SM, Farajzadeh MA, Hassanyani A, Afshar Mogaddam MR. Combination of homogenous liquid–liquid extraction and dispersive liquid–liquid microextraction for extraction and preconcentration of amantadine from biological samples followed by its indirect determination by flame atomic absorption spectrometry. RSC Adv. 2016;6:108603–10.
Somenath M. Sample preparation techniques in analytical chemistry. Hoboken: John Wiley & Sons Inc.; 2003. p. 227.
Altundag H, Tuzen M. Comparison of dry, wet and microwave digestion methods for the multi element determination in some dried fruit samples by ICP-OES. Food Chem Toxicol. 2011;49:2800–7.
Stolov AA, Kamalova DI, Borisover MD, Solomonov BN, Remizov AB. Hydrogen bonds formed by methyl groups of acetonitrile: Infrared and calorimetric study. Spectrochim Acta Part B. 1994;50:145–50.
Soylak M, Koksal M. Deep eutectic solvent microextraction of lead(II), cobalt(II), nickel(II) and manganese(II) ions for the separation and preconcentration in some oil samples from Turkey prior to their microsampling flame atomic absorption spectrometric determination. Microchem J. 2019;147:832–7.
Sorouraddin SM, Farajzadeh MA, Ghorbani M. In situ produced CO2 assisted dispersive liquid-liquid microextraction for extraction and preconcentration of cobalt, nickel, and copper ions from aqueous samples followed by graphite furnace atomic absorption spectrometry determination. J Iran Chem Soc. 2018;15:201–9.
Altunay N, Elik A, Gurkan R. Vortex assisted-ionic liquid based dispersive liquid liquid microextraction of low levels of nickel and cobalt in chocolate-based samples and their determination by FAAS. Microchem J. 2019;147:277–85.
Altunay N, Elik A, Bulutlu C, Gürkan R. Application of simple, fast and eco-friendly ultrasound assisted-cloud point extraction for pre-concentration of zinc, nickel and cobalt from foods and vegetables prior to their flame atomic absorption spectrometric determinations. Int J Environ Anal Chem. 2018;98:655–75.
Habibiyan A, Ezoddin M, Lamei N, Abdi K, Amini M, Ghazi-khansari M. Ultrasonic assisted switchable solvent based on liquid phase microextraction combined with micro sample injection flame atomic absorption spectrometry for determination of some heavy metals in water, urine and tea infusion samples. J Mol Liq. 2017;242:492–6.
Acknowledgments
The authors thank the Research Council of the University of Tabriz for financial support.
Funding
Saeed Mohammad Sorouraddin has received research grants from the University of Tabriz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects.
Informed consent
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 229 KB)
Rights and permissions
About this article

Cite this article
Okhravi, T., Sorouraddin, S.M., Farajzadeh, M.A. et al. Development of a liquid-nitrogen-induced homogeneous liquid–liquid microextraction of Co(II) and Ni(II) from water and fruit juice samples followed by atomic absorption spectrometry detection. Anal Bioanal Chem 412, 1675–1684 (2020). https://doi.org/10.1007/s00216-020-02406-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-02406-0