
Abstract
Reversible protein acetylation catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs) is an essential post-translational modification (PTM) mechanism which correlates largely with epigenetic gene regulation such as transcriptional activation, DNA replication, histone deposition, and DNA repair. Dysfunction of histone acetylation and the aberrant activity of HATs/HDACs is often associated with the pathogenesis of numerous diseases, especially cancer. Therefore, developing potent and specific analytical methods for HATs/HDACs is important for fundamental biochemical research, disease diagnosis and treatment, and drug development. This paper briefly summarizes the general design strategies used in HAT/HDAC sensors, gives a systematic overview of recent advances in the analytical methods for HAT/HDAC enzymatic analysis, classifies these methods by their biorecognition mechanisms and relative applications either in vitro or in living cells, then outlines challenges faced by these bioanalytical methods and offers perspectives on future developments.

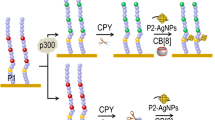
Reversible acetylation modification process and the general sensing mechanisms of protein acetylation-related enzymes (PAREs) activity
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reversible protein acetylation on lysine residues is one of the major post-translational regulatory mechanisms of protein activity, which is most extensively studied in histones and has long been the most prominent epigenetic modification that determines DNA accessibility and transcriptional activity [1, 2]. In particular, the level of histone lysine acetylation is controlled by the antagonistic catalytic activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), leading to the tight regulation of gene expression through chromatin modification [3]. Specifically, by acetylating histones, HATs reduce the net positive charge of histones, and thereby weaken electrostatic interactions with the negatively charged DNA, producing an open chromatin structure and resulting in greater accessibility of regulatory proteins to DNA. In particular, acetylation at multiple lysine residues is associated with decompaction of chromatin, providing space for the transcription machinery to engage with acetylated chromatin domains. HDACs, by contrast, catalyze acetyl group removal, leading to a closed chromosomal configuration and transcriptional repression.
Notably, dynamic lysine acetylation of histones is a typical “histone code” in the epigenetic marker system, which constitutes one of the most investigated and prominent epigenetic modification mechanisms [3]. Also, the recent discovery of many other lysine-acetylated proteins suggests that lysine acetylation is involved in diverse biological processes in cells, such as metabolism, cell cycle, splicing, nuclear transport, and actin nucleation [4]. Moreover, mounting evidence indicates that aberrant expression and dysregulation of HATs/HDACs is implicated in the etiology of numerous disease states [5, 6] including cancer, neurological disorders, chronic inflammation, and HIV infection; consequently, HATs/HDACs have attracted substantial attention and have emerged as important drug targets. Therefore, activity assays of protein acetylation-related enzymes (PAREs,including HATs and HDACs) are not only essential for the fundamental academic investigation on acetylation mechanism and function but also for acetylation-related pharmaceutical discovery. Also, selective small-molecule inhibition of PAREs presents novel routes for therapeutic intervention. To date, inhibitors of HDAC (HDACi) have been extensively studied and achieved notable clinical success. More than 20 HDACi are under different stages of preclinical and clinical development as single agents and in combination therapies against different cancers [7]. Zolinza (vorinostat), Istodax (romidepsin), and Beleodaq (belinostat) are three such drugs approved by the US Food and Drug Administration (FDA) for the treatment of rare T cell lymphoma [8].
Nevertheless, the screening and evaluation of HAT/HDAC-targeted inhibitors with potential pharmacological and therapeutic value is critically dependent on the availability of simple, sensitive, homologous, and robust quantitative assays of HATs/HDACs. Although several excellent reviews on HAT/HDAC families and their functions are available [9, 10], few focus on routine methods for PARE activity assays. Inspired by the significant development of acetylation-related medical and pathological research and the great advances in sensing methodology for HATs/HDACs, here we briefly review the recent, practical approaches for sensing HATs/HDACs that have not been covered by previous reviews, systematically classify these methods according to the biorecognition mechanism and application background, then highlight some excellent biosensors applicable for both extracellular and intracellular PARE analysis and discuss their relative advantages and limitations. Consequently, the challenges faced by these analysis methods are summarized and the possible directions for the future are discussed, which will hopefully provide some comprehensive and meaningful information for developing more novel PARE biosensors and promoting comparative drug screening studies.
Biosensing methods to detect PAREs in vitro
Protein acetylation is a reversible modification which involves covalent addition and removal of acetyl groups and requires the participation of co-reactants such as acetyl coenzyme A (AcCoA) and nicotinamide adenine dinucleotide (NAD+) (Fig. 1). Considering the variety and character of the products as well as the nature of the enzyme itself, plenty of biochemical assay technologies have been developed to evaluate the enzyme activity of HATs/HDACs. Radiometric assays were the traditional methods developed for PAREs, but these suffered from intrinsic hazards of radioactive materials and intricate multistep procedures. Hence some innovative non-isotopic methods were developed as alternatives, as shown in Fig. 1. Most of these assays were based on the direct identification of acetylated product to realize precise sensing, dependent on either the biorecognition by the specific proteins, including acetylation-specific antibodies and protein domains, or the variation of the physicochemical characteristics of acetylated products. Moreover, alternative methods based on the indirect quantitation of by-products such as coenzyme A (CoA) were established. Additionally, the high affinity binding between enzymes and their specific inhibitors provided a novel principle to detect HATs/HDACs or screen efficient inhibitors.

Reversible acetylation modification process and the general sensing mechanisms of PAREs activity
Acetyl-lysine-specific antibody-dependent assays
Depending on acetylation-specific antibodies, in combination with various fluorescence/colorimetric/electrochemical technologies, a series of immunoassays have emerged. Actually, Western blot and enzyme-linked immunosorbent assay (ELISA) were the conventional techniques for HATs/HDACs [11] and form the basis on which many commercial kits were designed. Yet this traditional method required multistep washing and separation processes, which made the assay relatively inconvenient, time-consuming, and low throughput. Thus in the past decade, especially with the rapid development of nanotechnology, numerous advanced nanomaterials were gradually introduced to realize homogeneous sensing of HAT/HDAC activity; this approach not only simplified the procedure and improved the throughput but also enhanced the selectivity, sensitivity, and reproducibility of the sensors.
Stevens et al. reported an interesting quantum dot (QD)-based HAT-sensing configuration via antibody recognition (Fig. 2a) [12]. After acetylation of the hexahistidine (His-tag)-appended synthetic substrate peptide, the acetylated peptide could then bind to the ZnS QD (emission maximum at 605 nm) surface by His-tag-mediated metal-affinity self-assembly; subsequent interaction with the Alexa Fluor 647-labeled acetyl-lysine-specific antibody resulted in effective fluorescence resonance energy transfer (FRET) from QDs to the Alexa Fluor 647, thus providing a simple and convenient optical readout for the acetylation process.

Antibody-dependent PAREs assays. a QD-based FRET sensor for detection of HAT activity. (Reprinted with permission from Ref. [12]; Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim) b Antibody-mediated assembly of AuNPs for colorimetric HAT sensing. (Reprinted with permission from Ref. [13]; Copyright 2012 American Chemical Society)
Jiang et al. developed a novel colorimetric biosensing strategy for activity screening of HAT (Fig. 2b) [13]. In this approach, gold nanoparticles (AuNPs) were first functionalized with the peptide substrates. Then after the HAT-catalyzed acetylation process, the anti-acetylpeptide antibodies recognized the acetylated peptides and their bivalent binding triggered the aggregation of AuNPs. This method represented the first example of using AuNP-based assembly for detecting HAT, which is homogeneous, simple, and versatile for other modifications by changing the corresponding antibody and peptide substrate.
The same group also developed a sensitive electrochemical biosensor for activity detection of sirtuin2 (SIRT2), a NAD-dependent class III HDAC [14]. The acetylated peptide-modified electrode could be recognized by the specific antibody and then coupled by secondary antibody conjugated with alkaline phosphatase (AP). The product of AP catalysis, naphthol, caused an amplified electrochemical signal. In the presence of SIRT2, the substrate was deacetylated, resulting in a decreased electrochemical signal that was correlated to the concentration of SIRT2.
Yu et al. developed a fluorescence immunoassay for the activity of SIRT2 utilizing an acetylated fluorescent peptide and its corresponding antibody-functionalized AuNPs [15]. The specific recognition between the acetylated lysine and the antibody led to the formation of an immunocomplex and efficient fluorescent quenching of fluorescent dyes by AuNPs. Enzymatic deacetylation of the peptide substrate directly weakened the interaction and could be detected by monitoring the fluorescence restoration of the sensing system, which was proportional to the activity of HDAC.
Antibody-free assays via recognition of acetylated products
Acetylation of lysine affords acetyl-lysine, which inevitably results in the biological and physicochemical properties of the substrate peptide changing significantly, such as the resistance to protease hydrolysis, the charge properties, or nucleophilicity. Rational design of detection mechanisms to discriminate the acetylation-induced change of these characteristics provides an efficient way to develop antibody-free methods for detecting PAREs.
Detection based on inhibition of protease degradation
It has been demonstrated that lysine acetylation greatly hinders the digestion of some proteases among which trypsin was the earliest studied [16, 17]. Trypsin cuts off the amide bond of lysine residues in the polypeptide chain and it is greatly hindered by the lysine acetylation. On the basis on this knowledge, researchers coupled the HDAC-catalyzed deacetylation with trypsin digestion to establish some primitive but frequently used bioassays both in laboratory and commercial assays. Schwienhorst et al. synthesized a new fluorogenic peptide substrate of HDACs with an acetylated lysyl moiety and an adjacent fluorophore moiety (7-amino-4-methylcoumarin, AMC) at the C terminus of the peptide chain [16, 17]. After deacetylation, the substrates became sensitive to trypsin and then induced the release of highly fluorescent AMC for subsequent detection. On the basis of a similar trypsin-resistant mechanism, Marcotte et al. then devised an acetylated peptide substrate doubly labeled with fluorescent tetramethylrhodamine-6-carboxylic acid (6-TAMRA) and quenching (QSY-7) groups for fluorescent turn-on SIRT assay [18].
Schwarzer et al. established a colorimetric detection for HDAC activities based on the 5-amino-2-nitrobenzoic acid (5,2-ANB)-tagged peptide substrate [19]. Furthermore, Ueki et al. exploited a standard HDAC assay with the substrate Boc-Lys(Ac)-AMC to measure the HDAC activity of a panel of cancer and normal cell lines. They observed marked high levels of HDAC activity in all malignant cell lines tested, which provided a selective environment for targeted cancer therapy [20]. Intriguingly, inspired by the ingenious structure of the probe, they coupled an acetylated lysine group to puromycin rather than AMC, creating a masked cytotoxic agent targeting increased HDAC and cysteine protease cathepsin L (CTSL) activities in malignant tumors. Then the activated puromycin selectively killed human cancer cell lines with high HDAC and CTSL activities. The cancer-selective cleavage of the small-molecule masking group may serve as a promising strategy for the development of the next generation of anticancer drugs.
Recently, Zhao et al. designed a specific turn-on fluorescent probe by connecting a specifically recognized peptide to a tetraphenylethene (TPE) core, which was capable of deacetylation-responsive aggregation-induced emission (AIE) for SIRT1 modulator screening and living cell imaging [21]. The fluorescent probe was non-emissive in aqueous medium. However, after deacetylation by SIRT1 and the subsequent digestion by lysyl endopeptidase, the N-terminal peptide of lysine was removed, and the hydrophobic TPE residues then aggregated together accompanied by the fluorescence emission in the aqueous solution; this approach was used to screen SIRT1 inhibitors and activators and track the expression of nucleus SIRT1 and its translocation in situ.
Our group developed an antibody-free fluorescent nanosensor to detect HAT activity based on the exopeptidase (CPY) digestion and the magnetic graphitic nanocapsule (MGN) as a novel nanoquencher (Fig. 3a) [22]. We first validated the effective suppression of CPY digestion by peptide acetylation, and utilized it as the recognition mechanism for HAT biosensor design. The lysine acetylation protected the fluorescein isothiocyanate (FITC)-tagged peptide probe against enzymatic cleavage and retained the peptide fragment capable of binding on the nanoquencher surface to form nanoquencher–peptide nanocomplexes, resulting in the fluorescence quenching. However, the unacetylated peptide is fully degraded by exopeptidase to release the fluorophore and restore fluorescence. The nanosensor presented a new sensing platform for probing HAT activity, with a low detection limit down to 0.1 nM and with a wide linear range from 0.5 to 100 nM.

Antibody-free PARE assays via recognizing the acetylated peptides. a HAT activity biosensor based on acetylation protection against CPY digestion and nanoquenchers. (Reprinted with permission from Ref. [22]; Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim) b Time-resolved luminescent biosensor for detection of PAREs activity based on the acetylation-mediated DNA–peptide interaction and the guanine-rich ssDNA-sensitized Tb3+ probes. (Reprinted with permission from Ref. [25]; Copyright 2015 American Chemical Society) c Peptide biosensor for one-step detection of HDAC activity via intramolecular transesterification. (Reprinted with permission from Ref. [29]; Copyright 2012 American Chemical Society) d Peptide-templated AuNCs as a label-free sensor for HAT activity. (Reprinted with permission from Ref. [32]; Copyright 2013 American Chemical Society)
Detection based on acetylation-induced peptide charge change
Acetylation eliminates the positive charge of the lysine residue and the acetylation or deacetylation-induced peptide charge alternation constitutes a relatively common HAT/HDAC detection principle. Kikuchi et al. synthesized a new TPE-derived fluorescent probe, K(Ac)PS-TPE, for the one-step detection of SIRT1 activity [23]. The probe was weakly fluorescent in solution and the deacetylation of lysine triggered the electrostatic interaction between its anionic sulfonate group and cationic lysine group. Thus, intermolecular aggregation occurred and automatically led to fluorescence enhancement based on AIE. The advantage of this probe was that the enzymatic activity was distinctly detected in a one-step procedure by simply mixing the probe and enzyme.
In a step further, the same group developed an alternative approach for probing HDAC activity on the basis of HDAC-regulated peptide charge change via converting neutral acetylated lysine to positively charged lysine [24]. They designed a DNA-based fluorogenic probe, BOXTO-GK(Ac)G, which was a DNA-staining dye–peptide conjugate containing an acetylated lysine for the detection of the activity of HDACs. After deacetylation, the DNA-dependent fluorescence of BOXTO-GK(Ac)G was greatly enhanced owing to the increased positive charge of the probe and its corresponding DNA binding ability, enabling the HDAC activity to be monitored in real time with a single-step manipulation.
Recently, our group proposed a label-free time-resolved luminescence (TRL) biosensor for real-time detection of enzymatic activity of HATs and HDACs (Fig. 3b), respectively, based on acetylation-mediated peptide/DNA interaction and Tb3+/DNA luminescent probes [25]. The acetylation of the peptide probe significantly altered its intrinsic charges and subsequently regulated its capability to bind with guanine(G)-rich ssDNA and replace Tb3+, resulting in the significant change of ssDNA-sensitized Tb3+ luminescence signals to reflect enzymatic activity of HATs or HDACs. With this TRL sensor, HAT (p300) can be sensitively detected with a wide linear range from 0.2 to 100 nM and a low detection limit of 0.05 nM. Additionally, it was successfully applied to continuously monitor HDAC (SIRT1) with a linear range from 0.5 to 500 nM and a detection limit of 0.5 nM. The proposed sensor was a convenient, sensitive, and mix-and-read assay, presenting a promising platform for protein acetylation-targeted epigenetic research and drug discovery.
Detection based on nucleophilicity alteration of lysine residues
Deacetylation is accompanied by the transformation from acetylated imine to primary amine on a lysine residue, causing the evident increase of nitrogen nucleophilicity and thus presents another potent recognition mechanism for HDAC. Early in 2003, Jung et al. presented several HDAC probes that relied on the fluorescent substrate Boc(Ac)Lys-AMC, also termed MAL or Z-MAL [26, 27]. After incubation of the substrate with HDAC, a nucleophilic derivatization applying the classical amine derivatization agent naphthalene-2,3-dialdehyde (NDA) led to efficient fluorescence quenching of the deacetylated metabolite. Thus, only the fluorescence emitted by the remaining substrate was detected, which allowed for the homogeneous detection of HDAC activity.
However, as these assays require multistep and laborious procedures, Kikuchi et al. developed a single-step continuous HDAC assay through intramolecular nucleophilic derivative reactions (Fig. 3c) [28, 29]. The fluorogenic probe, K4(Ac)-CCB, consisted of the histone H3 peptide with acetyl-lysine and a coumarin fluorophore with an electrophilic carbonate ester in the 7-hydroxy group as a fluorogenic switch responsive to the deacetylation reaction. By simple incubation of the probe with HDAC, enzyme activity was clearly detected through spontaneous intramolecular transesterification, which switched the probe fluorescent signal on.
Recently, Buccella et al. reported a new small molecular probe for HDAC activity which contained an acetyl-lysine mimic functionalized with an amine-reactive coumarin fluorophore and this served as both HDAC substrate and optical reporter [30]. This new probe was designed to undergo rapid intramolecular condensation reaction between aldehyde and amine upon deacetylation-induced nucleophilic change. The process led to a bathochromic shift in the absorption spectrum and evident changes in fluorescence intensity that enabled real-time detection of HDAC activity of purified enzymes or in cell lysates.
Other mechanisms to recognize acetylated products
There are also several examples of special sensing mechanisms which are noteworthy. Zheng and Wu designed a series of peptide-based HAT reporters; the change in photophysical properties after acetylation was exploited to provide a homogeneous, continuous, and one-step method of reporting HAT activity [31]. Jiang et al. reported that peptide-templated fluorescent gold nanoclusters (AuNCs) retained the substrate activity and that enzymatic modifications, such as acetylation, of the peptides were able to quench the fluorescence of the AuNCs; this effect was attributed to the peptide acetylation probably affecting the compactness of the protective coating and inducing O2-mediated fluorescence quenching of the AuNCs (Fig. 3d). On the basis of this, they developed a real-time and label-free sensing strategy for HDAC1 [32].
Li et al. designed an electrochemical strategy for the evaluation of the effect of p53 C-terminal domain (CTD) acetylation on DNA binding ability. They first immobilized peptides derived from the CTD of p53 with single or multiple acetylated lysine residues on electrode surfaces. Then, ferrocene (Fc)-labeled dsDNA probe was introduced to bind with the p53 CTD and give different electrochemical responses corresponding to the acetylation degree. The results showed that acetylation of p53 CTD enhanced its DNA binding ability which was positively correlated with the acetylated lysine residue numbers. This electrochemical method provides a simple method to study p53 protein acetylation [33].
Moreover, Hof et al. reported a simple chemical sensor array that relied on a mix-and-match toolkit of several available dyes and calixarene host molecules for antibody-free reading of the histone code [34]. The sensors were based on an indicator displacement scheme in which a fluorescent dye was bound to and quenched by a suitable host molecule. Addition of acetyl-lysine could compete for the host’s binding site and cause the release of some or all of the dyes, thus resulting in a fluorescent turn-on response, and the fluorescence intensity change was unique to the concentrations and identities of the histone modifications. Although this method did not achieve the direct assay of PAREs, the supramolecular recognition mechanism shows great potential for further PARE biosensor development.
Determination of coproduct generation
Acetylation reaction involves the acetyl group transfer from AcCoA to lysine residues on the substrate peptide, producing an acetylated peptide and CoA coproduct. Probing the generation of CoA is a straightforward way to detect HAT activity. One approach relies on enzymatic coupling reaction. Denu et al. developed two continuous assays for the prototypical GCN5 HAT [35]. In these assays, the HAT-catalyzed acetylation was continuously coupled with an enzymatic system containing either α-ketoglutarate or pyruvate dehydrogenase. The CoA generated in the HAT reactions was measured by accompanied reduction of NAD+ to NADH spectrophotometrically in the sequential enzymatic reaction.
On the other hand, CoA is a thiol compound composed of cysteine, pantothenate, and adenosine groups. Since CoA contains a thiol group, the direct quantitation of CoA by thiol-responsive fluorescent probes is a facile approach. Marmorstein et al. developed a sensitive fluorometric HAT assay by quantifying the production of CoA in the enzymatic turnover through direct reaction with a sulfhydryl-sensitive fluorophore, 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) [36]. Zheng et al. then systematically evaluated a series of such thiol-sensitive fluorogenic compounds for different HAT activity assays [37].
Nevertheless, the selectivity of the aforementioned fluorescent reporters of CoA might be problematic because they are specific to only the thiol group rather than intact CoA. Because the structure of CoA contains both a thiol group and adenine base, our group proposed a novel fluorescent assay for probing HAT activity based on in situ generation of a nucleic acid-mimicking coordination polymer (CP) of CoA–Au(I), which utilized CoA as an organic ligand and exhibited high specificity for CoA [38]. Notably and interestingly, we found that, as a nanochain-like polymer with multiple adenine bases as the side groups, CoA–Au(I) CP is likely analogous to the structure of poly-A single-strained nucleic acid and can combine with the RNA-selective dye SYBR Green II (SGII) and emit strong fluorescence, thus presenting a promising mechanism for one-step HAT activity assay and inhibitor screening (Fig. 4a). Furthermore, we prepared another nucleic-acid mimicking CoA–Ag(I) CP, which can effectively bind with graphene oxide (GO) through its adenine side groups via π–π stacking interaction and present unexpected high electrocatalytic activity for H2O2 reduction. This principle has been used to develop a novel, label-free electrochemical sensor to probe HAT activity (Fig. 4b) [39].

HAT assays based on the coproduct CoA determination. a Fluorescent HAT biosensor based on CoA–Au(I) coordination polymer (CP) stained by SGII. (Reprinted with permission from Ref. [38]; Copyright 2015 The Royal Society of Chemistry) b The electrochemical biosensor for probing HAT activity dependent on the electrocatalytic activity of CoA–Ag(I) CP loaded on graphene oxide (GO). (Reprinted with permission from Ref. [39]; Copyright 2015 The Royal Society of Chemistry)
Competition binding assays based on PARE inhibitors
Researchers have also exploited the selective interaction between HAT/HDAC and their specific inhibitors to develop a series of competition binding assays. Although these methods cannot directly reflect the enzymatic activity, they actually provide a simple and alternative platform to screen the PARE inhibitors. Meyer-Almes et al. demonstrated a competition assay for HDACi using the HDAC homologue (HDAH) and 2-furylacryloylhydroxamate (FAHA), a new fluorescent HDACi, which was based on the FRET from tryptophans of HDAH to FAHA upon its binding to the enzyme [40]. The same group applied a similar competition binding mechanism to develop another homologous assay of HDAC in which two parameters were affected [41], namely fluorescence anisotropy and fluorescence lifetime. This assay possesses the advantages of one-step measurement coupled with robustness against autofluorescent compounds and it is therefore highly suitable for high-throughput screening of specific inhibitors of HDAH and HDACs. Zheng et al. also proposed another dual-mode fluorometric strategy for screening HAT modulators by both FRET and anisotropy measurements [42]. In the sensor design, HATs were labeled with Dabcyl (Dab) as FRET acceptors and methoxycoumarin (Mca) was conjugated to HAT substrate analogues to function as fluorescent donors. When HAT interacted with the substrate analogues to form a complex between the acceptor-labeled HAT protein and the donor-labeled ligand, the fluorescent intensity of the donor fluorophore decreased as a result of FRET quenching by the Dab acceptor. The formation of ligand–protein complexes also caused reduction of the molecular mobility of the donor fluorophore, resulting in increased fluorescence anisotropy. This allowed for simultaneous fluorescence intensity and anisotropy readout and enabled facile evaluation of inhibitor potency without extra specific substrates.
Detection of PAREs activity and protein acetylation level in living cells
Most of the traditional HAT and HDAC assays are applicable for in vitro enzymatic analysis of PAREs, whereas monitoring the dynamic changes of acetylation level and PAREs activity in single cells and living organism is still highly challenging. To address this issue, several cell-based assays were developed to measure overall acetylation level and the enzymatic activity in living cell systems. Some of the pioneering work on cell-based assaying technologies are discussed in this section.
Bromodomains, an extensive family of evolutionarily conserved protein modules, have long been known to function as the intrinsic acetyl-lysine-binding recognition domains in vivo and were found in many chromatin-associated proteins, including HATs such as PCAF and TAF1, and the BET family of nuclear proteins, including BRD2, BRD3, BRD4, and BRDT [43]. Bromodomains can “read” specific acetylation events “written” by HATs, thus providing bridges for protein–protein interactions, which serves as a pivotal mechanism for epigenetic regulation of chromatin remodeling and transcriptional activity via lysine acetylation [44]. On the basis of these, several FRET-based probes were built to evaluate the levels of protein acetylation or the balance between HATs and HDACs in living cells (Fig. 5a). These FRET-based probes were typically tandem fusion proteins consisting of a substrate, a flexible linker, a bromodomain recognizing the acetylated site, and two different colored mutants of green fluorescence protein (GFP), for example, cyan fluorescent protein (CFP) and yellow fluorescent protein (Venus). Upon acetylation, the substrate and bromodomain bind to each other and cause an intramolecular structural change that alters the distance and orientation of the two fluorescent proteins to disturb FRET. Early in 2004, Ozato et al. reported the first FRET-based analysis combined with flow cytometry (termed FC-FRET) to evaluate highly selective recognition between different acetylated histones and bromodomain-containing proteins in living cells [45].

Cellular acetylation level evaluation. a Genetically encoded FRET biosensor to monitor histone acetylation based on the acetylation-specific bromodomain. (Reprinted with permission from Ref. [49]; Copyright 2015 American Chemical Society) b Visualization of histone acetylation level in living cell using Fab-based endogenous modification labeling. (Reprinted with permission from Ref. [50]; Copyright 2011 Oxford University Press)
The first successful application of FRET-based imaging probe in recognizing acetylated histone prompted researchers to further develop more advanced probes for specific acetylation sites. To examine when, where, and how histone acetylation is “written” or “erased” by HATs or HDACs in living cells, Yoshida et al. developed several novel bromodomain-based imaging tools for site-specific histone modifications [46–49]. The first example was the chromatin-targeted probe named Histac, which was designed for visualizing the dynamic fluctuation of H4K5/K8 acetylation during mitosis [46] and the action of a p300/CBP inhibitor [47]. Then the Histac-K12 probe was used to detect cellular histone H4K12 acetylation while characterizing a small-molecule modulator of acetylation or interaction status of histones [48]. Recently, the same group developed another FRET probe to monitor the acetylation of histone H3 on K9/K14 based on its specific binding to BRD4 [49].
Another kind of cellular protein acetylation level assay is based on fluorescently labeled acetylation-specific antigen binding fragments (Fabs), prepared from purified mouse IgG by protease digestion. Kimura et al. demonstrated a general method to monitor the spatial gradients and global level of endogenous histone H3 lysine modifications in single living cells without disturbing cell growth and embryo development [50]. Once being loaded into the cytoplasm of living cells, Fabs can enter the nucleus and produce distinct nuclear patterns that were characteristic of their target modifications. As Fabs bind their targets transiently, the ratio of bound and free Fabs depended on the concentration of acetylation sites, allowing us to monitor rapid changes in global modification levels by simply comparing the concentration of Fabs in the nucleus and cytoplasm (Fig. 5b). However, this method requires direct loading of Fabs into cells, and their dilution upon cell division prevents long-term and organism-level experiments. The same group further described a genetically encoded system that enables the tracking of histone H3 lysine 9 acetylation (H3K9Ac) continuously [51]. Specifically, they fused a single-chain variable fragment (scFv) antibody to the enhanced green fluorescent protein (EGFP) to create a modification-specific intracellular antibody, or ‘mintbody’, which retained high specificity for H3K9 acetylation and demonstrated broad potential for tracking histone modification in vivo.
Outlook
We have summarized the current advances in the development of HAT/HDAC biosensors and bioassays for bioanalysis, biochemistry, and drug screening research. Although remarkable progress has been achieved in enzymatic analysis of acetylation-related enzymes, significant challenges still exist. Firstly, since the majority of assays are discontinuous, a potential goal is to develop continuous assays for HATs/HDACs capable of real-time monitoring of the acetylation/deacetylation process, which is important for enzyme kinetics analysis. Secondly, some of the existing biosensors have achieved the detection of HAT/HDAC activity in cell lysates, but monitoring HAT/HDAC-catalyzed acetylation in intracellular environment is still challenging. Endocellular or even in situ detection will provide valuable information for understanding the function of HATs/HDACs in the epigenetics as well as inhibitor-related drug screening in living systems. Thirdly, another promising trend is to introduce some novel nanomaterials with appealing characters, such as NIR QDs or upconversion materials, to enhance the specificity and the sensitivity of the assay probes, which enables the detection in the NIR region for in vivo imaging. Developing robust strategies for efficient delivery of these nanoprobes into living cells and specifically locating them in the cell nucleus are also significant for intracellular HAT/HDAC studies. To sum up, the efforts addressing the above goals will eventually produce fruitful chemical tools that can strengthen our understanding of the complex role of HATs/HDACs in health and disease a step further.
References
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80.
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327(5968):1000–4.
Biel M, Wascholowski V, Giannis A. Epigenetics-an epicenter of gene regulation: histones and histone-modifying enzymes. Angew Chem Int Ed. 2005;44(21):3186–216.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40.
Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14(19):942–8.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–91.
Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4(4):505–24.
Ratner M. Small biotech steers HDAC inhibitor to clinic. Nat Biotechnol. 2014;32(9):853–4.
Yang XJ, Seto EY. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–8.
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400.
Kuninger D, Lundblad J, Semirale A, Rotwein P. A non-isotopic in vitro assay for histone acetylation. J Biotechnol. 2007;131(3):253–60.
Ghadiali JE, Lowe SB, Stevens MM. Quantum-dot-based FRET detection of histone acetyltransferase activity. Angew Chem Int Ed. 2011;123(15):3479–82.
Zhen Z, Tang L, Long H, Jiang J. Enzymatic immuno-assembly of gold nanoparticles for visualized activity screening of histone-modifying enzymes. Anal Chem. 2012;84(8):3614–20.
Wu H, Liu S, Zhu W, Jiang J, Shen G, Yu R. A sensitive electrochemical biosensor for detection of histone deacetylase activity using an acetylated peptide. Electroanalysis. 2012;24(12):2365–70.
Zhang S, Wu H, Huan S, Zhang X, Shen G, Yu R. Gold nanoparticle based fluorescence resonance energy transfer immunoassay for the detection of the histone deacetylase activity using a fluorescent peptide probe. Anal Lett. 2013;46(13):2029–39.
Wegener D, Hildmann C, Riester D, Schwienhorst A. Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal Biochem. 2003;321(2):202–8.
Wegener D, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol. 2003;10(1):61–8.
Marcotte PA, Richardson PL, Guo J, Barrett LW, Xu N, Gunasekera A, et al. Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Anal Biochem. 2004;332(1):90–9.
Dose A, Jost JO, Spiess AC, Henklein P, Beyermann M, Schwarzer D. Facile synthesis of colorimetric histone deacetylase substrates. Chem Commun. 2012;48(76):9525–7.
Ueki N, Lee S, Sampson NS, Hayman MJ. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat Commun. 2013;4(4):2735.
Wang Y, Chen Y, Wang H, Cheng Y, Zhao X. Specific turn-on fluorescent probe with aggregation-induced emission characteristics for SIRT1 modulator screening and living-cell imaging. Anal Chem. 2015;87(10):5046–9.
Han Y, Li P, Xu Y, Li H, Song Z, Nie Z, et al. Fluorescent nanosensor for probing histone acetyltransferase activity based on acetylation protection and magnetic graphitic nanocapsules. Small. 2015;11(7):877–85.
Dhara K, Hori Y, Baba R, Kikuchi K. A fluorescent probe for detection of histone deacetylase activity based on aggregation-induced emission. Chem Commun. 2012;48(94):11534–6.
Minoshima M, Matsumoto T, Kikuchi K. Development of a fluorogenic probe based on a DNA staining dye for continuous monitoring of the histone deacetylase reaction. Anal Chem. 2014;86(15):7925–30.
Han Y, Li H, Hu Y, Li P, Wang H, Nie Z, et al. Time-resolved luminescence biosensor for continuous activity detection of protein acetylation-related enzymes based on DNA-sensitized terbium(III) probes. Anal Chem. 2015;87(18):9179–85.
Heltweg B, Dequiedt F, Verdin E, Jung M. Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal Biochem. 2003;319(1):42–8.
Heltweg B, Jung M. A homogeneous nonisotopic histone deacetylase activity assay. J Biomol Screen. 2003;8(1):89–95.
Baba R, Hori Y, Kikuchi K. Intramolecular long-distance nucleophilic reactions as a rapid fluorogenic switch applicable to the detection of enzymatic activity. Chem Eur J. 2015;21(12):4695–702.
Baba R, Hori Y, Mizukami S, Kikuchi K. Development of a fluorogenic probe with a transesterification switch for detection of histone deacetylase activity. J Am Chem Soc. 2012;134(35):14310–3.
Rooker DR, Buccella D. Real-time detection of histone deacetylase activity with a small molecule fluorescent and spectrophotometric probe. Chem Sci. 2015. doi:10.1039/c5sc02704g.
Wu J, Zheng YG. Fluorescent reporters of the histone acetyltransferase. Anal Biochem. 2008;380(1):106–10.
Wen Q, Gu Y, Tang L, Yu R, Jiang J. Peptide-templated gold nanocluster beacon as a sensitive, label-free sensor for protein post-translational modification enzymes. Anal Chem. 2013;85(24):11681–5.
Zheng D, Ye Z, Yang S, Li Y, He X, Li G. An electrochemical method to evaluate p53 C-terminal domain acetylation on its DNA binding ability. Electrochem Commun. 2014;49:30–3.
Minaker SA, Daze KD, Ma MC, Hof F. Antibody-free reading of the histone code using a simple chemical sensor array. J Am Chem Soc. 2012;134(28):11674–80.
Kim Y, Tanner KG, Denu JM. A continuous, nonradioactive assay for histone acetyltransferases. Anal Biochem. 2000;280(2):308–14.
Trievel RC, Li FY, Marmorstein R. Application of a fluorescent histone acetyltransferase assay to probe the substrate specificity of the human p300/CBP-associated factor. Anal Biochem. 2000;287(2):319–28.
Gao T, Yang C, Zheng YG. Comparative studies of thiol-sensitive fluorogenic probes for HAT assays. Anal Bioanal Chem. 2013;405(4):1361–71.
Chen S, Li Y, Hu Y, Han Y, Huang Y, Nie Z, et al. Nucleic acid-mimicking coordination polymer for label-free fluorescent activity assay of histone acetyltransferases. Chem Commun. 2015;51(21):4469–72.
Hu Y, Chen S, Han Y, Chen H, Wang Q, Nie Z, et al. Unique electrocatalytic activity of nucleic acid-mimicking coordination polymer for sensitive detection of coenzyme A and histone acetyltransferase activity. Chem Commun. 2015. doi:10.1039/c5cc06593c.
Riester D, Hildmann C, Schwienhorst A, Meyer-Almes FJ. Histone deacetylase inhibitor assay based on fluorescence resonance energy transfer. Anal Biochem. 2007;362(1):136–41.
Riester D, Hildmann C, Haus P, Galetovic A, Schober A, Schwienhorst A, et al. Non-isotopic dual parameter competition assay suitable for high-throughput screening of histone deacetylases. Bioorg Med Chem Lett. 2009;19(13):3651–6.
Xie N, Elangwe EN, Asher S, Zheng YG. A dual-mode fluorescence strategy for screening HAT modulators. Bioconjug Chem. 2009;20(2):360–6.
Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–6.
Yang X. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays. 2004;26(10):1076–87.
Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell. 2004;13(1):33–43.
Sasaki K, Ito T, Nishino N, Khochbin S, Yoshida M. Real-time imaging of histone H4 hyperacetylation in living cells. Proc Natl Acad Sci U S A. 2009;106(38):16257–62.
Dancy BM, Crump NT, Peterson DJ, Mukherjee C, Bowers EM, Ahn YH, et al. Live-cell studies of p300/CBP histone acetyltransferase activity and inhibition. Chem Biol Chem. 2012;13(14):2113–21.
Ito T, Umehara T, Sasaki K, Nakamura Y, Nishino N, Terada T, et al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem Biol. 2011;18(4):495–507.
Nakaoka S, Sasaki K, Ito A, Nakao Y, Yoshida M. A genetically encoded FRET probe to detect intranucleosomal histone H3K9 or H3K14 acetylation using BRD4, a BET family member. ACS Chem Biol. 2015. doi:10.1021/cb501046t.
Hayashi-Takanaka Y, Yamagata K, Wakayama T, Stasevich TJ, Kainuma T, Tsurimoto T, et al. Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res. 2011;39(15):6475–88.
Sato Y, Mukai M, Ueda J, Muraki M, Stasevich TJ, Horikoshi N, et al. Genetically encoded system to track histone modification in vivo. Sci Rep. 2013;3:2436.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos. 21222507, 21175036, 21235002, and 21475038), the National Basic Research Program of China (973 Program, No. 2011CB911002), the Foundation for Innovative Research Groups of NSFC (Grant 21221003), and the Natural Science Foundation of Hunan Province (No. 2015JJ1005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no potential conflict of interest.
Additional information
Published in the topical collection featuring Young Investigators in Analytical and Bioanalytical Science with guest editors S. Daunert, A. Baeumner, S. Deo, J. Ruiz Encinar, and L. Zhang.
Rights and permissions
About this article

Cite this article
Li, P., Han, Y., Li, Y. et al. Bioanalytical approaches for the detection of protein acetylation-related enzymes. Anal Bioanal Chem 408, 2659–2668 (2016). https://doi.org/10.1007/s00216-016-9304-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9304-7