
Abstract
The purity determination of organic calibration standards using the traditional mass balance approach is described. Demonstrated examples highlight the potential for bias in each measurement and the need to implement an approach that provides a cross-check for each result, affording fit for purpose purity values in a timely and cost-effective manner. Chromatographic techniques such as gas chromatography with flame ionisation detection (GC-FID) and high-performance liquid chromatography with UV detection (HPLC-UV), combined with mass and NMR spectroscopy, provide a detailed impurity profile allowing an efficient conversion of chromatographic peak areas into relative mass fractions, generally avoiding the need to calibrate each impurity present. For samples analysed by GC-FID, a conservative measurement uncertainty budget is described, including a component to cover potential variations in the response of each unidentified impurity. An alternative approach is also detailed in which extensive purification eliminates the detector response factor issue, facilitating the certification of a super-pure calibration standard which can be used to quantify the main component in less-pure candidate materials. This latter approach is particularly useful when applying HPLC analysis with UV detection. Key to the success of this approach is the application of both qualitative and quantitative 1H NMR spectroscopy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A significant challenge facing analytical laboratories working in the field of chemical testing is the availability of high-quality calibration standards for use in quantitative analysis. Ideally, these calibration standards will have certified purity values with established traceability to the International System (SI) unit for mass (kg), ensuring comparability and reliability of chemical measurement worldwide [1, 2]. Within this context, it is imperative that reference material producers can maintain an efficient and cost-effective production process while simultaneously ensuring an appropriate level of certification is achieved, with particular attention being paid to the uncertainty requirements of the end-user.
Traditionally, the purity of so-called pure (or neat) substances has been determined using a suite of complementary analytical techniques. Gas chromatography (GC) and high-performance liquid chromatography (HPLC) provide a means of separating the impurities from the main component (usually the analyte of interest), and the use of a suitable detector facilitates their quantification. Flame ionisation detection (FID) interfaced with GC provides a measure of carbon, making it ideal for the analysis of volatile organic compounds [3–8]. Analytes more suited to liquid chromatography are detected and quantified by way of UV/Vis spectroscopy (UV), and for those not possessing a suitable chromophore, evaporative light scattering detector (ELSD) and charged aerosol detector (CAD) are applicable. Summation of all impurities (I ORG) and subtraction from 100 % provides a measure of what shall be termed the ‘organic purity’ in this paper.
The mass fraction of volatile impurities (I VOL), common organic solvents and/or water, can be evaluated by means of thermogravimetric analysis (TGA) at elevated temperatures, and Karl Fischer analysis provides a direct measure of water. Combustion or ‘ashing’ of the sample, at temperatures exceeding 600 °C, completes the mass balance approach [9–20] with a measure of the non-volatile residue (I NVR), usually assumed to be inorganic salts, in the sample. Summation of all volatile and non-volatile impurities and subtraction from 100 % completes the mass balance assessment of ‘total purity’ of a given material, which is calculated using Eq. 1. The purity value calculated as a percentage can then be readily converted into a mass fraction (mg/g).
The organic purity of a candidate material will invariably be assessed using the chromatographic technique most suited to the physical characteristics of the main analyte. Having established suitable chromatographic conditions, the next challenge is the conversion of peak areas into an accurate realisation of the relative mass fractions. One approach is to assume that all impurities are of similar structure to the main analyte, on the grounds that they have not been removed during extensive purification procedures (recrystallisation and/or column chromatography) [9, 18, 19]. It follows that each impurity can be expected to elicit a near-identical response to the main analyte, thereby justifying the use of peak areas as a direct measure of the relative mass fraction of each component. While this model no doubt holds true for many samples analysed by gas chromatography with flame ionisation detection (GC-FID), it is by no means failsafe and is particularly vulnerable when applied to samples analysed by high-performance liquid chromatography with UV detection (HPLC-UV) where subtle changes in structure can have a profound effect on the molar response. An alternative, metrologically robust approach, adopted in more recent times, is to identify each impurity and quantify via calibration [11, 17, 20], eliminating the potential for bias arising from the assumption described above. Unfortunately, this introduces the almost impossible task of securing calibration standards for each impurity, simultaneously increasing the time, and cost, associated with the certification process.
Ongoing experience analysing compounds of varying structural complexity, including steroids and steroid metabolites, illicit drugs, pharmaceuticals, agrochemicals and veterinary products, has highlighted the potential for significant bias in measurements made using thermogravimetric analysis and the need to, once again, challenge some of our initial assumptions.
Using examples to highlight the problems encountered over the past 10–15 years, this paper will detail the measures we have implemented to limit the impact of hidden bias. In the process, we have developed an approach which negates the need to quantify impurities of nominally similar structure to the main component via external calibration, leading to an efficient and practical certification protocol. Problems associated with the application of ELSD for purity determination have been highlighted in numerous publications and will not be discussed further in this paper [21].
Experimental
All weighings were performed on a calibrated Mettler Toledo XP205 or XS205 Dual Range five-figure analytical balance reading from 0.00001 to 220 g.
GC-FID analysis was performed using a 2000 μg/mL solution of the candidate material. Linearity has been demonstrated between 2000 and 0.4 μg/mL for a suite of analytes with carbon percentages in the range 50–80 %. Injection volume was 1 μL and split ratio typically 20:1. The carrier gas was helium at 1.0 mL/min. Injection temperature ranges from 180 to 250 °C. Columns typically employed include HP-1 (30 m × 0.33 mm × 0.25 μm in film thickness) and DB-5 (30 m × 0.33 mm × 0.25 μm in film thickness).
HPLC-UV/Vis analysis was performed using a Waters 1525 binary pump coupled to a Waters 2998 photodiode array detector, and Empower processing software. Samples were made up in the eluent at a concentration equivalent to the upper limit (μg/mL) of the linear range determined for the main analyte. All chromatography grade solvents were purchased from Merck (LiChrosolv®). Ultrapure (deionised) water was prepared with a Milli-Q Gradient system (Millipore SAS, Molsheim, France). Injection volume was 10 μL. The eluent flow rate was 1.0 mL/min. Columns typically employed include Ascentis, X-Bridge and Alltima (150 mm × 4.6 mm × 5.0 μm in particle size).
HPLC-UV/Vis analysis of avermectin B1a
Three Waters Symmetry C18 columns, 5 μm, 4.6 mm × 150 mm, and a Waters Symmetry C18 column, 5 μm, 3.9 mm × 150 mm, were connected in tandem. The mobile phase (20 % Milli-Q water/80 % acetonitrile) was run at a flow rate of 1 mL/min at ambient temperature.
LC-MS analysis of avermectin B1a
Three Waters Symmetry C18 columns, 5 μm, 4.6 mm × 150 mm, and a Waters Symmetry C18 column, 5 μm, 3.9 mm × 150 mm, were connected in tandem. The mobile phase (20 % Milli-Q water containing 1 % formic acid/80 % acetonitrile) was run at a flow rate of 1 mL/min at ambient temperature. Post chromatography, the flow rate was amended to 0.2 mL/min to the Waters 2695 Alliance Quattro MS, operating with MassLynx software. The following conditions were used: capillary voltage, 3.5 kV; cone voltage, 50 V; extractor voltage, 3 V; source temperature, 130 °C; de-solvation temperature, 350 °C; cone gas flow, 29 L/h; and de-solvation gas flow, 767 L/h.
Preparative HPLC-UV/Vis purification of avermectin B1a
Technical grade avermectin B1a was chromatographed over a Waters Symmetry C18 column, 19 mm × 150 mm × 7 μm, eluting with acetonitrile/Milli-Q water (80:20) at a flow rate of 9 mL/min. Fractions containing avermectin B1a (front and back fractions were discarded) were combined, and the organic solvent was removed under reduced pressure. The remaining cloudy aqueous solution was extracted with diethyl ether (3 × 100 mL), dried (Na2SO4) and evaporated in vacuo to afford an oil. This oil was dissolved in a minimum volume of ethyl acetate and treated with hexane to induce crystallisation. The resulting white powder was collected and dried under reduced pressure at 40 °C to yield a high-purity sample of avermectin B1a.
Karl Fischer analysis was performed on Mettler Toledo DL32 and Metrohm 756 coulometers. Hydranal®-Water standards 0.10 (0.1 mg/g) and 1.0 (1.0 mg/g) for Karl Fischer were purchased from Riedel-de Haen and Fluka Analytical, respectively. Lactose monohydrate obtained from Merck was used as the QC check sample.
Thermogravimetric analysis was performed using a Perkin Elmer Pyris 1 or Mettler Toledo TGA/DSC 1 instruments using 5–15 mg of sample. Typical temperature program was 25 °C (hold for 1.0 min), 25–120 °C at 40 °C/min (hold for 20 min), 120–850 °C at 100 °C/min (hold for 10 min) and 850–25 °C at 40 °C/min (hold for 10 min). In many cases, a higher temperature, up to 160 °C, and longer hold times may be necessary to drive off all occluded solvent.
1H NMR spectra were acquired on a Bruker Avance III spectrometer operating at 500.13 MHz, equipped with 5 mm BBFO probes, and B-ACS-60 sample changers, under the control of Bruker’s TopSpin software, version 3.1. Deuterated NMR solvents such as acetone-d 6 99.9 atom %, chloroform-d 99.8 atom %, deuterium oxide 99.9 atom %, dimethylsulfoxide-d 6 99.9 atom % and methanol-d 4 99.8 atom % were products of Cambridge Isotope Laboratories, Inc. All chloroform-d was stored over anhydrous potassium carbonate before use.
Results and discussion
Determination of organic purity
Our approach to organic purity determination is focused on avoiding the need to secure calibration standards to quantify each impurity. The first requirement is to establish that the main analyte (typically present at greater than 99 % purity) and all impurities are measured within the linear range of the detector. Once this has been addressed, we then need to assess the response factor of each component to facilitate an accurate conversion of chromatographic peak areas into relative mass fractions. Fortunately, the assessment of organic purity also presents an opportunity to evaluate the homogeneity of the sample, a key requirement in the certification of reference materials [22]. As a general rule, ten sub-samples will be selected from various sections of the sample and analysed in duplicate, simultaneously providing an assessment of organic purity and homogeneity, statistical analysis of variance (ANOVA) determining method repeatability (u wb) and between sub-sample inhomogeneity (u bb) [23, 24]. For each chromatographic technique discussed in this paper, this will form the core of the measurement uncertainty (MU) budget associated with the organic purity value. Additional uncertainty components will be discussed where relevant.
Gas chromatography with flame ionisation detection
For analytes amenable to gas chromatography, this is without doubt the preferred high-resolution separation technique, because of its relatively simple operation. Furthermore, high sensitivity, a broad linear range (typically 2000–0.4 μg/mL), and the expectation that compounds of similar structure (carbon percentage and functionality) will elicit a similar response, making the FID the detector of choice [3–8]. In our experience, the majority of samples have impurities eluting within ±1 min of the main analyte, supporting the assumption of ‘like structure’ which is in line with our original working model [9]. However, the observation of impurities with very different retention times to the main analyte prompted us to question the validity of this assumption and introduced the need to identify each impurity and assess the relative response factor. This led to the uncomfortable realisation that, on occasion, impurities of significantly different structure were found to elute close to the main analyte (Fig. 1).

The structures of key compounds discussed in the text
GC-FID analysis of the liberated free base of 4-methyl-α-pyrrolidinopropiophenone hydrochloride 1 provides an ideal case study. The major impurity was found to elute within a minute of the main analyte with a relative peak area of 0.5–0.6 %. GC-MS analysis confirmed a near-identical fragmentation pattern for both compounds, suggesting an isomeric relationship between the two. 1H NMR analysis of the hydrochloride salt confirmed the impurity to be the 3-methyl isomer, integration confirming a relative mass fraction of 0.5–0.6 % as per the GC-FID analysis. Unfortunately, a further ten impurities, all at peak areas below 0.05 %, could not be identified due to difficulties obtaining mass spectral data and/or confirming their presence in the 1H NMR. In the absence of structural information, we need to account for the fact that the structure/response factor of one or more unidentified impurities may be very different to the main analyte. This is achieved through the development of a conservative uncertainty component which can be added to the overall MU budget for the assigned purity value. Based on the knowledge that the dominant influence on individual response factors is the carbon percentage of the compound in question, we can postulate a worst case scenario for the conversion of peak areas into the relative mass fraction of unidentified components. Considering the synthetic route employed to make this material, it is extremely unlikely that any impurity will have a carbon percentage which differs by more than a factor of two compared to the main analyte (4-methyl-α-pyrrolidinopropiophenone has a carbon percentage of 77.4 %). Therefore, a worst case scenario can be envisaged in which the sum relative peak area of all ten impurities (0.28 %) could actually represent a relative mass fraction of 0.56 % (5.6 mg/g relative mass fraction). Assuming a rectangular distribution centred at the measured value and extending to the proposed upper limit allows us to calculate a conservative uncertainty (u RF = 0.28 %/√3 = 0.16 %) associated with the quantification of these impurities, simultaneously avoiding the need to calibrate each impurity (Fig. 2). Furthermore, this approach allows for the fact that none of these impurities have been identified in the 1H NMR, and may, in fact, be artefacts of the measurement process. This issue is dealt with in more details later in the text.

Model used to calculate the uncertainty associated with the conversion of GC-FID chromatographic peak areas of unidentified impurities into relative mass fractions. The uncertainty calculation is based on a rectangular probability distribution centred at the measured value (0.28 %) and a hypothetical upper mass fraction (0.56 %) based on carbon percentage of individual components
In stark contrast was our GC analysis of a sample of 5-iodo-2-aminoindane hydrochloride 2 for which varying levels of halogenation were found to have a dramatic impact on individual response factors. The chromatogram (see Electronic Supplementary Material (ESM) Fig. S1) shows the free base of 2 eluting at 6.4 min and a major impurity at 10.8 min with a peak area of 4.9 %. Based on GC-MS analysis (385 m/z base peak was observed) and the singlet at 7.88 ppm in the 1H NMR (Fig. 3), the major impurity was identified as the hydrochloride salt of 5,6-di-iodo-2-aminoindane 3, while a second impurity eluting at 8.61 min (~0.9 %) was identified as 5-chloro-6-iodo-2-aminoindane hydrochloride 4. The challenge of converting the peak areas into relative mass fractions for each hydrochloride salt is made all the more difficult by each free base having a significantly different carbon percentage and, hence, response factor in the FID, as well as each hydrochloride salt having a different free base to HCl ratio, all of which needs to be accounted for. To overcome this problem, the relative mass fractions of 2, 3 and 4 were determined by 1H NMR run under quantitative conditions [25–28]. Integration of the one-proton doublet at 7.10 ppm (main analyte 2), two-proton singlet at 7.88 ppm (impurity 3) and the one-proton singlet at 7.47 ppm (impurity 4) confirmed the relative mass fraction of the impurities to be 7.2 % (72 mg/g) and 1.1 % (11 mg/g), respectively, both values being significantly different to the relative peak area percentage by GC-FID, in line with expectations. While 1H NMR resolved the issues associated with GC-FID analysis, integration of individual peaks was not without its problems. The preferred practice of using integrals of sufficient width to include the corresponding 13C satellites was only possible with 2 (186 Hz integral width), integral widths of peaks representing 3 (89 Hz) and 4 (23 Hz) being significantly reduced to avoid interference with neighbouring peaks. Even so, the integral representing 3 at 7.88 ppm included a signal for 4 and the downfield 13C satellite from H4 of 2, which needed to be evaluated and subtracted from the integral value. Furthermore, both integrals representing 3 and 4 also needed to be corrected to account for the omission of the 13C satellites, representing 1.1 % of the overall peak area. To assess the effect that integral width has on the accuracy with which each component can be quantified, the one-proton doublet of 2 at 7.10 ppm was integrated using both 186 Hz (full integral) and 74 Hz integral widths. The full integral (assigned a value of 100) includes an impurity peak (triplet at 6.99 ppm) which accounts for 0.33 % of the total integral, 99.67 % being assigned to 2. An integral width of 74 Hz (value = 98.54) avoids interference with the impurity peak and, after correction for the omission of the 13C satellites, affords a total integral value of 99.63, confirming that the use of narrower integral widths has minimal impact on the assigned peak area. This may not hold so true for the even narrower integral of the one-proton singlet at 7.47 ppm representing impurity 4. Fortunately, this impurity is present at sufficiently low levels (1.1 %) that the impact of any error in the accuracy of this integral can be expected to be minimal.

1H NMR spectrum of a sample of 5-iodo-2-aminoindane 2 in MeOH-d 4, run under qNMR conditions (D 1 = 30 s, 256 scans), used to determine the relative mass fractions of impurities 3 and 4
Interestingly, correction of the GC-FID peak areas for impurities 3 and 4, based solely upon carbon percentage relative to 5-iodo-2-aminoindane 2, afforded peak area percentages of 1.0 and 7.0 %, respectively, in line with the quantification by 1H NMR, suggesting that carbon percentage is by far the dominant factor when considering relative response factors in the FID.
The two examples presented here demonstrate the value of cross-checking the relative mass fraction of impurities using an alternative technique [17], the most effective being 1H NMR spectroscopy acquired under quantitative conditions [25–27]. It is also clear that while unequivocal identification is desirable, it is by no means essential, spectroscopic data supporting similarity of structure being sufficient in most cases. For example, it was not essential to identify the major impurity in the sample of 4-methyl-α-pyrrolidinopropiophenone hydrochloride 1 as the 3-isomer, evidence of the isomeric relationship being sufficient. It should also be noted that this example represents the most commonly encountered scenario, in which most identifiable impurities have structures sufficiently similar to the main component that correction for response factor is not considered necessary, particularly when present at low levels (<1 %). In the event that impurities are present with significantly different structures to the main analyte and variations in carbon percentage/response factors cannot be corrected for by 1H NMR, then the analyst may consider developing individual uncertainty components in the manner described above.
During the course of ongoing testing, it has become apparent that some compounds display a propensity to degrade upon injection onto the silanised glass liner. This is not too surprising, considering the need for elevated temperatures (180–250 °C) to volatilise the analyte onto the GC column. The most dramatic example of this effect to date has been observed with a sample of 17-epioxandrolone 5. Three impurity peaks eluting within a minute of 5 were observed, one consistently at 0.5 % relative peak area, while the other two were shown to vary between 0.2–0.8 and 0.1–0.4 % depending on the injection port used for the analysis. Demonstrated absence of multiple impurities between 0.4 and 0.8 % mole fraction in the 1H NMR supports the hypothesis that the variability of the two impurities is the result of degradation of the main component, although in the absence of supporting information, it would be prudent to also consider the possibility that these impurities are actually degrading into the main analyte.
In the majority of cases, ‘liner sensitivity’ is not always so obvious. Our initial analysis of a sample of testosterone 6 afforded an organic purity of 99.2 % with a standard deviation of 0.03 % (n = 10). It was only when an annual stability trial was performed did the potential for degradation, albeit by only 0.4 %, become apparent (see Fig. 4). Our ability to identify the issue in the first instance was hampered by two outcomes. Firstly, the data set was very precise, wrongly suggesting that the analysis was bias free, and secondly, the two chromatographic peaks later identified as artefacts of the injection process were only present in 0.2–0.3 %, making it difficult to confirm their presence or absence in the relatively complex 1H NMR spectrum of 6. With further data available, it is clear that the mean organic purity estimates obtained after the initial analysis are more accurate representations of the true organic purity value. No two values differ by more than 0.2 %, although Student’s t test analysis between the latter four data sets suggests some of the means to be statistically non-equivalent at the 95 % confidence interval [23].

Summary of organic purity estimates of a sample of testosterone by GC-FID
To circumvent this problem, we have implemented a simple, but effective safeguard of analysing each new candidate material using two or three different injection ports to evaluate the effect of liner quality. In practice, very rarely are the mean values of any two organic purity determinations statistically equivalent, and in the absence of further evidence (1H NMR and/or HPLC-UV/Vis) to reject one or more data sets, each is considered a suitable measure of organic purity and combined to afford an overall mean and standard deviation. One outcome of this approach is a larger standard deviation (u SD), although for the majority of our calibration standards, this is rarely a cause for concern. In line with international guidelines, homogeneity is determined from ANOVA of one data set only [24].
While the modified approach provides greater confidence in the measure of organic purity, it was noted in the above example that the artefact peaks are still present in at least 0.05 %, which raises the question as to whether the highest estimations of organic purity are also biased to some small degree or these two impurities are actually present in the sample, their relative percentage being increased through degradation on other occasions. This can be dealt with most efficiently by treating the impurity as unknown in the manner described before for 4-methyl-α-pyrrolidinopropiophenone hydrochloride 1.
In summary, the combined standard uncertainty associated with organic purity determinations by GC-FID is summarised in Eq. 2.
High-performance liquid chromatography with UV detection
The organic purity of polar and/or thermally labile compounds is most conveniently assessed using HPLC with UV/Vis detection, generally, but not exclusively, performed at the λ max of the main analyte [11]. The linear range will be determined on a case-by-case basis, and the analysis performed at the upper limit, assuming the absence of solubility issues. Ideally, all components will possess the same chromophore, chromatographic peak areas representing the relative molar ratio, which, in turn, can be converted into the relative mass fraction using the respective molecular weights. In reality, this is a rare situation, subtle changes in structure often having a significant impact on the molar response (extinction co-efficient/molar absorptivity) of individual components. The previously reported analysis of testosterone sulfate 7 demonstrated how the mass fraction of one impurity, displaying a significantly different λ max, was identified by a combination of LC-MS and 1H NMR spectroscopy, and quantified by the latter [28]. 1H NMR analysis on multiple sub-samples, ideally under quantitative conditions, will afford a mean molar ratio (MRNMR) for the impurity in question, which can then be used to calculate a correction factor (CF) for the representative HPLC peak area. Combining the standard deviation associated with the NMR analysis (s NMR) and the standard deviation associated with the HPLC analysis (s HPLC), as described in Eq. 3, will provide an overall uncertainty associated with the correction factor (u CF) for a given impurity.
In the event that limited material is available, an alternative approach is to perform 1H NMR analysis on a single sub-sample utilising as many peaks associated with the main analyte and impurity as possible, multiple measures of the molar ratio of the impurity of interest provide the required mean and standard deviation.
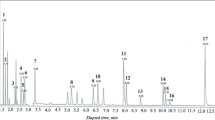
Participation in an international comparison study into the purity assessment of avermectin B1a 8 presented a somewhat greater challenge. Optimised chromatographic conditions successfully separated the expected impurities [29] from the main analyte (Fig. 5) as well as a tentatively assigned diastereomer of avermectin B1a (rt = 26.9 min) which we believe to be the first time this has been reported. Apart from avermectin B1b which was identified as the main impurity at 21.5 min, all other impurities were characterised by UV/Vis and electrospray ionisation mass spectrometry (ESI-MS) as summarised in Table 1. Six of the eight main impurities displayed a characteristic λ max at 245 nm with shoulders at higher and lower wavelengths (ESM Fig. S2) in line with that observed for avermectin B1a and B1b. Accordingly, the chromatographic trace was processed at 245 nm, and consideration of the respective molecular weights of all identified components afforded an organic purity of 94.2 % (942 mg/g relative mass fraction) for the avermectin B1a. Karl Fischer analysis confirmed the presence of 30.2 mg/g water, affording a total purity value of 913 ± 12 mg/g.

HPLC-UV/Vis chromatogram of a sample of avermectin B1a analysed at 245 nm
In the process of performing the analysis, a number of concerns were raised. Firstly, ESI-MS analysis confirmed that the first peak to elute from the column (15.1 min) represented the co-elution of three compounds tentatively assigned as an isomer of avermectin B1b (858.2 m/z), hydrogenated avermectin B1a (874.5 m/z) and hydrated avermectin B1a (890.6 m/z). Strictly speaking, a lack of information regarding the molar ratio of the three compounds precludes an accurate conversion to mass fractions, although in reality, use of the highest (890.6 m/z) and lowest (858.2 m/z) molecular weights in the calculation made no difference to the total purity value.
Also of concern was the fact that the impurities eluting at 16.5 and 41.6 min did not display the characteristic UV spectrum of all the other components, with λ max at 249 and 243 nm, respectively, without the characteristic shoulders, suggesting a minor change in the conjugation along the butadiene portion of the molecule, and the potential for an associated change in the molar extinction coefficients of these impurities compared to the other components, which, in turn, precludes direct conversion of peak areas into relative molar ratios. The third concern was related to the fact that the approach does not account for any impurities present which lack the appropriate chromophore. While the concerns raised above were expected to be of minor consequence, we still felt that a suitable cross-check for bias should be put in place. Unfortunately, the number of impurities in the sample and the complexity of the 1H NMR spectrum made it impossible to accurately cross-check the organic purity value of this sample using 1H NMR spectroscopy, as detailed above. To address these concerns, we subjected a technical grade sample to extensive purification by preparative HPLC to afford a sample free of all impurities except the diastereomer at 26.9 min. This sample was certified for purity, a process made all the easier by the fact that the response factor issue had been eliminated, facilitating facile determination of the organic purity, and subsequently used to quantify the avermectin B1a 8 in the test sample. This afforded a purity estimate of 915 ± 12 mg/g, which compared favourably to our original assessment of 913 ± 8 mg/g, Student’s t test confirming that the two independently determined purity values are statistically equivalent [P(T > t) = 73 %] at the 95 % confidence interval [23]. The successful approach detailed here follows the lead of Le Goff and Wood [11]. The significant investment of time and resources to create higher-purity calibration standards, ideally 100 % pure, will be offset by the ease with which batches of lower organic purity can be certified, circumventing the need to determine the response factor of one or more impurities.
Determination of volatile and non-volatile residue content
The assessment of volatile content, primarily organic solvents and water, in a given material, has traditionally been performed by thermogravimetric analysis and water-specific Karl Fischer analysis. In cases where the only volatile present is water, the results from both analyses should match perfectly, providing confidence in both measurements. Lactose monohydrate is one such example, liberating 51 mg/g of moisture at 160 °C in line with expectations, and is used routinely as the QC sample for both techniques.
Full combustion of the test sample at temperatures exceeding 600 °C facilitates a measure of the non-volatile residue. For the majority of samples, the residue is below the limit of detection, determined to be 2 mg/g mass fraction, in line with expectations for most organic compounds. In these circumstances, the mass fraction of non-volatile residue was assigned as zero and the associated uncertainty was calculated, assuming a rectangular probability distribution between the two limits, zero and 2 mg/g.
Thermogravimetric analysis
Of prime importance when conducting thermogravimetric analysis is confirming that the observed change in mass provides an accurate measure of total volatiles. Temperatures between 120 and 160 °C are generally considered sufficient to drive off all volatile content, although it is by no means guaranteed, as demonstrated by our analysis of 6-monoacetyl morphine 9. Heating to 160 °C resulted in a weight loss of 1–2 mg/g which was confirmed to be biased by the observation of 5 mg/g of water by Karl Fischer analysis. Scrutiny of the 1H NMR (ESM Fig. S3) confirmed further bias in the TGA result with both dichloromethane and diethyl ether being present in 9 and 7 mg/g mass fraction, respectively. This example serves, once again, to demonstrate the importance of using 1H NMR spectroscopy to cross-check the results of more traditional techniques used in purity determination. Conscious of the fact that solvent content will not always be so readily identified, we have introduced a qualitative headspace GC-MS analysis to our suite of characterisation techniques. This is particularly useful when the preparation history of the candidate material is unknown.
Certifying a range of steroid glucuronides isolated, for convenience, as the sodium salt, introduced a problem when assessing non-volatile residue content. Combustion of the sample afforded a residue of sodium salts of undefined stoichiometry, preventing an accurate measure of sodium ions in the sample. This was particularly important in this example as we were keen to determine if surplus sodium was present in the form of other salts, e.g. sodium hydroxide, or, indeed, if there was a deficiency of sodium ions, other cations or the free acid comprising part of the sample. In these circumstances, inductively coupled plasma–optical emission spectroscopy offers a solution to the problem, providing a direct analysis of sodium, though large sample size requirements and relatively large uncertainties (10–20 %) prohibit the use of this technique, particularly when months of synthesis work have delivered less than 200 mg of product. One approach to overcome this problem would be to change the measurand to the steroid glucuronate anion and certify the purity using qNMR [25–28], thereby ignoring the presence of sodium or any other counter ions in the sample. However, in most cases, this problem has been overcome by converting the sodium salt into the free acid, a relatively simple process in which an aqueous solution of the sample was titrated to pH 2, precipitating the free acid thereby eliminating the problem altogether.
Karl Fischer water analysis
A surprisingly small number of compounds in our collection are water free, making Karl Fischer analysis a crucial component to the mass balance approach. This viewpoint has been reinforced by the observation that absorption of water is the major cause of change in purity. In light of the previously detailed failure to liberate total volatiles, including water, at elevated temperatures, we prefer direct addition of the sample to the coulometric titration cell rather than using the Karl Fischer oven method. However, this approach also introduces a number of issues. Firstly, upon addition of the sample, the coulometric cell is exposed to ambient moisture. This can be accounted for by running a series of blanks, in which the transfer process is replicated in the absence of sample. The corresponding mean is subtracted from the mass of water determined for a given test sample to afford the blank corrected value. Accordingly, the associated uncertainty (\( {u}_{\mathrm{Corrected}\ {\mathrm{H}}_2\mathrm{O}} \)) is calculated by combining the standard deviation (\( {s}_{\mathrm{Blank}\ {\mathrm{H}}_2\mathrm{O}} \)) of n blank measurements and the uncertainty (\( {u}_{\mathrm{Raw}\ {\mathrm{H}}_2\mathrm{O}} \)) associated with a given measure of water, as shown in Eq. 4. Unfortunately, the consumption of each sub-sample upon analysis prevents an assessment of \( {u}_{\mathrm{Raw}\ {\mathrm{H}}_2\mathrm{O}} \), essentially method repeatability, in the manner described for the chromatographic techniques, i.e. via duplicate analysis. To address this problem, we have exhaustively analysed a homogeneous sample of lactose monohydrate to afford a relative standard deviation for the certified mass fraction of water (51 mg/g). Based on the assumption that this is applicable to all levels of water, the relative standard deviation is used to estimate the \( {u}_{\mathrm{Raw}\ {\mathrm{H}}_2\mathrm{O}} \) for the measured mass of water in a given sub-sample.
The mass fraction of water for a given test sample is calculated from the blank corrected value and the sample mass, the uncertainty associated with the latter coming from the balance calibration. The overall uncertainty (\( {u}_{{\mathrm{H}}_2\mathrm{O}} \)) associated with a given measure of water, as a mass fraction percentage (H2O%), is calculated using Eq. 5.
Two further components are incorporated into the MU budget, the standard deviation of n test samples and a bias component, the latter being determined from the difference between the mean water content determined for three to four sub-samples of lactose monohydrate and the certified water content of 51 mg/g.
Karl Fischer titrations can be performed in either Hydranal® AG or Hydranal® AK reagent solutions, depending on solubility and functionality of the analyte of interest. Samples of Hydranal® Water Standard 1.0, containing 1 mg H2O per g, were analysed in both solutions and found to afford mean mass fraction values that can be considered to be statistically equivalent as judged using Student’s t test [P(T > t) = 39 %] at the 95 % confidence interval [23]. This alleviated concerns that the previously reported comparatively slow and sluggish titration in 2-methoxyethanol-based Hydranal® AK reagent solution was having a detrimental effect on the outcome of the titration compared to the favoured methanolic Hydranal® AG solution [30]. Hydranal® AK reagent solution is primarily used for the coulometric titration of aldehydes and ketones when there is a need to suppress the formation of acetals and ketals and water as a by-product. A dramatic demonstration of this effect was seen with a sample of 6β-hydroxyturinabol 10, Karl Fischer analysis affording a measure of 63 mg/g water in AG solution and 27 mg/g in AK. Supporting evidence for the AK result came from thermogravimetric analysis at 160 °C, affording a measure of total volatiles at 22 mg/g. Conscious of the aforementioned potential for bias in the thermogravimetric result, we thought it prudent to obtain alternative confirmation of the AK result. This was provided by elemental microanalysis which confirmed a carbon percentage of 66.2 and 7.3 % hydrogen in the sample, which is more consistent with a water content of 27 mg/g (calculated C = 66.6 %, H = 7.9 %) than the 63 mg/g (C = 64.2 %, H = 8.0 %). Furthermore, based on the acceptance criteria of ±0.3 % used in most organic chemistry journals, the microanalysis suggests the presence of impurities other than water and, hence, the need for further analysis.
Conclusions
The mass balance approach described provides a fit for purpose assessment for organic calibration standards providing purity values which are traceable to the SI unit for mass (kg). In the case of Karl Fischer results, the traceability to the kilogram is established via traceability to another SI unit, the ampere (A). While ideally suited to compounds of high total purity (>990 mg/g), the approach is equally applicable to materials of far lower purity. Key to the success of the approach is the ability to cross-check each measurement result using an independent technique, for which demonstrated equivalence using statistical tools such as Student’s t test provides greater confidence in the assigned purity value. The quantification of impurities of similar structure to the main analyte using chromatographic techniques falls into three categories. Demonstrated equivalence of response to the main analyte in the detector of choice facilitates the direct conversion of peak areas into relative mass fractions, negating the need for calibration studies. In cases where impurities with different response factors to the main analyte are present, the relative mass fraction can be determined, in most cases, using 1H NMR spectroscopy acquired under quantitative conditions. Alternatively, it may be possible to use a different chromatographic technique. The third scenario deals with impurities for which it is not possible to get a positive identification and, hence, assessment of response in the detector of choice. For samples analysed by GC-FID analysis, potential bias arising from significant variation in molecular structure can readily be accommodated by application of a conservative measurement uncertainty budget based on worst case scenarios for carbon percentages in the unidentified impurities compared to the main analyte. In a perfect scenario, the provision of candidate materials devoid of impurities of similar structure eliminates the need to determine response factors, simultaneously removing the potential for bias in the quantification. Time invested to furnish such high-purity materials will be compensated by the relative ease of certification as well as the long-term ability to quantitate the same analyte in candidate materials of lower purity.
Ultimately, despite the many safeguards implemented to cross-check each measurement result, it is not possible to completely rule out the potential for hidden bias using the mass balance approach alone, reaffirming the value of quantitative NMR to directly measure the analyte of interest, thereby providing the best available cross-check [28]. However, the analyst needs to be mindful of the fact that poor resolution of an impurity in both the NMR and chromatography may lead to a situation in which the mass balance and qNMR determined purity values match perfectly belying the presence of hidden bias. This scenario is increasingly likely as the analytes of interest increase in size and complexity, and variations in the structure of the impurities are minor, the sample of avermectin B1a providing a perfect example.
References
De Bievre P, Taylor PDP (1997) Metrologia 34:67
De Bievre P, Taylor PDP (1993) Int J Environ Anal Chem 52:1
Hill HH, McMinn DG (1992) Detectors for capillary chromatography, 1st edn. Wiley-Interscience
Perkins G, Laramy RE, Lively LD (1963) Anal Chem 35(3):360–362
Edwards RWH (1978) J Chromatogr 153:1–6
Tong HY, Karasek FW (1984) Anal Chem 56:2124–2128
Jorgensen AD, Picel KC, Stamoudis VC (1990) Anal Chem 62:683–689
Yieru H, Quinyu O, Weile Y (1990) Anal Chem 62:2063–2064
King B, Westwood S (2001) Fresenius J Anal Chem 370:194–199
Duewer DL, Parris RM, White E, May WE, Elbaum H. (2004) An approach to the metrologically sound traceable assessment of the chemical purity of organic reference materials. NIST Special Publication 1012; NIST: Gaithersburg, MD
Le Goff T, Wood S (2008) Anal Bioanal Chem 391:2035–2045
Le Goff T, Wood S (2009) Anal Bioanal Chem 394:2183–2192
Westwood S, Daireaux A, Josephs RD, Wielgosz RI et al (2009) Metrologia 46(1A):08019
Westwood S, Daireaux A, Josephs RD, Wielgosz RI et al (2011) Metrologia 48(1A):08013
Westwood S, Daireaux A, Josephs RD, Wielgosz RI et al (2012) Metrologia 49(1A):08009
Westwood S, Daireaux A, Josephs RD, Wielgosz RI et al (2012) Metrologia 49(1A):08014
Ishikawa K, Hanari N, Shimizu Y, Ihara T, Nomura A, Numata M, Yarita T, Kato K, Chiba K (2011) Accred Qual Assur 16:311–322
Yip Y-C, Wong S-K, Choi S-M (2011) Trends Anal Chem 30(4):628–640
Kim S-H, Lee J, Ahn S, Song Y-S, Kim D-K, Kim B (2013) Bull Kor Chem Soc 34(2):531–538
Westwood S, Choteau T, Josephs RD, Daireaux A, Wielgosz RI (2013) Anal Chem 85:3118–3126
Mojsiewicz-Pienkowska K (2009) Crit Rev Anal Chem 39:89–94
ISO Guide 34 (2009) General requirements for the competence of reference material producers, 3rd edn, clause 3.5. International Organisation for Standardisation (ISO), Geneva
Hibbert DB, Gooding JJ (2006) In data analysis for chemistry: an introductory guide for students and laboratory chemists. Oxford University Press, Oxford
ISO Guide 35 (2006 E) Reference materials—general and statistical principles for certification, 3rd edn. International Organisation for Standardisation (ISO), Geneva
Al-Deen TS Ph.D. Thesis, University of NSW, submitted September 2002. Validation of quantitative nuclear magnetic resonance (QNMR) spectroscopy as a primary ratio analytical method for assessing the purity of organic compounds: a metrological approach
Malz F, Jancke HJ (2005) J Pharm Biomed Anal 38(5):813–823
Saito T, Ihara T, Miura T, Yamada Y, Chiba K (2011) Accred Qual Assur 16:421–428
Davies SR, Jones K, Goldys A, Alamgir M, Chan BKH, Elgindy C, Mitchell PSR, Tarrant GJ, Krishnaswami MR, Luo YW, Moawad M, Lawes D, Hook JM (2015) Anal Bioanal Chem 407(11):3103–3113
Awasthi A, Razzak M, Al-Kassas R, Harvey J, Garg S (2012) Chem Pharm Bull 60(8):931–944
Scholz E (1984) Karl Fischer titration. Springer-Verlag, Berlin Heidelberg New York
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 193 kb)
Rights and permissions
About this article

Cite this article
Davies, S.R., Alamgir, M., Chan, B.K.H. et al. The development of an efficient mass balance approach for the purity assignment of organic calibration standards. Anal Bioanal Chem 407, 7983–7993 (2015). https://doi.org/10.1007/s00216-015-8971-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8971-0