
Abstract
A simple and fast method for analysis of hydroxylated polycyclic aromatic hydrocarbons using pressurized liquid extraction and high performance liquid chromatography utilizing photoionization tandem mass spectrometry was developed. Simultaneous separation and determination of nine hydroxylated polycyclic aromatic hydrocarbons and two hydroxy biphenyls could be performed in negative mode with a run time of 12 min, including equilibration in 5 min. The calibration curves were in two concentration ranges; 1–50 ng/mL and 0.01–50 μg/mL, with coefficients of correlation R 2 > 0.997. The limits of detection and method quantification limits were in the range of 9–56 pg and 5–38 ng/g, respectively. A two-level full factorial experimental design was used for screening of conditions with the highest impact on the extraction. The extraction procedure was automated and suitable for a large number of samples. The extraction recoveries ranged from 70 to 102 % and the matrix effects were between 92 and 104 %. The overall method was demonstrated on wood smoke particles and soil samples with good analytical performance, and five OH-PAHs were determined in the concentration range of 0.19–210 μg/g. As far as we know, hydroxylated polycyclic aromatic hydrocarbons were determined in wood smoke and soil samples using photoionization mass spectrometry for the first time in this present study. Accordingly, this study shows that high performance liquid chromatography photoionization tandem mass spectrometry can be a good option for the determination of hydroxylated polycyclic aromatic hydrocarbons in complex environmental samples.

The method developed in this study was used to determine hydroxylated polycyclic aromatic hydrocarbons in wood smoke and soil
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are environmental contaminants with carcinogenic properties [1–4] and are mainly formed from anthropogenic sources in incomplete combustion processes [5, 6]. Hydroxylated PAHs (OH-PAHs) are formed by microbiological and/or photochemical degradation of PAHs in the urban environment, and in the body as metabolites of PAHs [7, 8]. Unlike PAHs and the oxy- and nitro-substituted PAHs, OH-PAHs have not been studied as extensively. It has, however, been shown that OH-PAHs are potentially more toxic and carcinogenic than the native PAHs [9–12]. Apart from mutagenicity, OH-PAHs are also endocrine disruptors. Among many others, 1-hydroxy pyrene (1-HP), 2- and 3-hydroxy phenanthrene (2-HPh, 3-HPh), and 2-hydroxy chrysene have been shown to have estrogenic activity [13–15], whereas e.g., 3-hydroxy benzo[a]pyrene (3-HBaP) has been shown to have strong anti-estrogenic activity [14, 15]. A large part of the research done on OH-PAHs has focused on them as metabolites of PAHs in biological samples [16, 17].
There have been difficulties to detect OH-PAHs attributable to matrix effects and complicated sample pretreatments. Both high performance liquid chromatography (HPLC) and gas chromatography mass spectrometry (GC-MS) have been used [18–21]. Sample preparation for GC analysis is, however, time-consuming because of a necessary derivatization step since OH-PAHs are polar, thermally unstable compounds with a tendency to form hydrogen bonds. Solid-phase microextraction (SPME) with chemical derivatization has been used for analysis of PAH metabolites in human urine [22–24], but the method is not suitable when analyzing a large number of samples. A couple of LC-MS methods have been reported for determination of OH-PAHs using atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI) [25–30]. Another ionization technique, atmospheric pressure photoionization (APPI) is available for ionization of nonpolar and semipolar molecules and has been shown to be more suitable for analysis of OH-PAHs than ESI [31]. In a study, APPI was shown to be more sensitive towards all test compounds (carbamazepine, acridine, naphthalene, diphenyl sulphide, and 5-fluorouracil) compared with APCI [32]. APPI is also more tolerant of matrix components that can interfere with the mechanisms in ESI and APCI [33] and has successfully been used for determination of PAHs and nitro-substituted PAHs in complex environmental matrices [34].
As stated above, most of the currently available methods for determination of OH-PAHs are based on GC-MS, requiring a derivatization step or HPLC-MS often based on ESI suffering from severe matrix effects when analyzing complex matrices. However, HPLC-MS is better suited for analysis of polar compounds because of easy sample pretreatments, short run times, and softer ionization. Since there have been few studies in the literature on the application of APPI to environmental analysis, although it has been shown to be more tolerant to matrix effects, the aim of the present study has been to develop a simple, fast, and sensitive analytical method for simultaneous determination of nine OH-PAHs and two hydroxy biphenyls in different environmental samples, using pressurized liquid extraction (PLE) and HPLC-APPI-MS/MS. In order to enable fast and sensitive detection of trace amounts of OH-PAHs in complex matrices, the different steps of the analytical procedure such as the extraction, clean-up, separation, and ionization have been evaluated. The suitability of the developed method has then been illustrated by application to wood smoke particles and soil samples. In contrast to PAHs, only few studies are available determining OH-PAHs in urban soils [35] and wood smoke particles [36], while it has been shown that the ambient aerosols contain significant amounts of OH-PAHs [37]. The analytes have been chosen based on their toxicity and occurrence in the matrices analyzed, but also because some of the selected analytes are formed by hydroxylation of frequently recorded PAHs in PAH-contaminated environments, e.g., naphthalene, phenanthrene, chrysene, and pyrene.
Materials and methods
Chemicals and solvents
2-Hydroxy biphenyl (2-HBP; 98 %) was purchased from Fine Chemicals AB (Uppsala, Sweden) and 4-hydroxy biphenyl (4-HBP; 98 %) from Merck-Schuchardt (Hohenbrunn, Germany). 2-Hydroxy-9-fluorenone (2-H-9-F; 98)% was from Sigma-Aldrich (Milwaukee, WI, USA), 1-HP (98 %) from Sigma-Aldrich (Steinheim, Germany), 1,8-dihydroxy anthraquinone (1,8-DHAQ; 96 %) from Sigma-Aldrich (Shanghai, China), 1-naphthol (1-N; ≥99 %) from Sigma-Aldrich (Buchs, Switzerland), and 2-naphtol (2-N; 99 %) from Sigma-Aldrich (Milan, Italy). 3-HPh (99 %) and 6-hydroxy chrysene (6-HC; 99 %) were from Dr. Ehrenstorfer GmbH (Augsburg, Germany), 2-HPh (99 %) and 3-HBaP (98 %) from Neochema (Bodenheim, Germany), and deuterated 1-hydroxy pyrene (1-HP-d9; 99 %) was from Chiron AS (Trondheim, Norway). 1-HP-d9 has similar chemical and psychical properties as 1-HP and was used as internal standard (IS) for all of the analytes. It was used for evaluation of the recoveries in the different extraction and clean-up steps. More details on the analytes and IS are shown in Table 1.
Methanol, toluene, and acetonitrile (ACN) were of HPLC grade and were purchased from Rathburn Chemicals Ltd. (Walkerburn, UK). Anisole (99 %) was from Merck-Schuchardt (Hohenbrunn, Germany) and ammonium acetate (analysis grade) was from Merck (Darmstadt, Germany). Water was purified using a Millipore Synergy 185 (Millipore Corp., Danvers, MA, USA) water purification system equipped with a Millipak 0.22 μm membrane filter (Millipore).
Stock solutions with concentrations of approximately 0.1 g/L in methanol were prepared for all compounds, except the analytes 2-HPh, 3-HPh, and 3-HBaP, which were purchased as solutions in acetonitrile with the concentrations of 0.1 g/L. Working standard solutions were prepared from the stock solutions by diluting 100 μL of each stock solution with methanol to a final volume of 1 mL. The stock solutions of each analyte were stored in a freezer (–20 °C) and were stable for at least 1 y.
Samples
Wood smoke particles
Wood smoke particles were generated using a conventional natural draft wood stove, fired with birch logs with a moisture content of 16–17 %. The wood stove was operated in a laboratory flue gas setup, including a flue gas fan to ensure a relevant chimney draft throughout the combustion cycles. The stove was operated in two modes; nominal burn rate (NB) with “normal” combustion conditions, and high burn rate (HB) with high load intensive combustion. During the combustion, approximately 2 and 3 kg/h of wood logs (dry mass) were consumed in the different modes, respectively. During the NB mode, the stove maintained a rather good combustion (CO in the range 500–5000 ppm) throughout the burn-cycle, whereas during the HB mode the high burn rate caused short episodes of “flash over” with oxygen supply deficiency (CO in the range 1000–20,000 ppm), in accordance with a previous study [38].
The emission measurements were performed in the flue gas channel, approximately 1.5 m above the stove at flue gas temperature of 100 − 300 °C, depending on combustion conditions. The raw flue gases were diluted around four times by a porous tube diluter [39] operated with HEPA filtered dilution air at ambient temperature. The wood smoke particles were sampled on MK360, 90-mm micro-quartz fiber filters (Munktell Filter AB, Falun, Sweden). The filters were conditioned in a desiccator and weighed on an analytical balance (scale accuracy of ±0.01 mg) before and after the sampling and stored in a freezer (–20 °C) until chemical analysis. The combustion and sampling set-ups and procedures are described in detail elsewhere [38, 40]. Three wood smoke samples were analyzed in the present study; wood smoke sample 1 (HB case) and wood smoke sample 2 and 3 (NB cases). Blank filters were also generated and analyzed.
Soil samples
The soil samples were collected at the industrial area of Värtahamnen in Stockholm, Sweden, which contains an industrial area adjacent to a harbor for large cargo and commercial ships. The area, in particular, is of interest because of the combustion of fossil fuels from ships and the industries. The following three sampling locations were chosen: the harbor terminal (59°21.010′N 18°6.333′E) as sampling location 1, an industrial area owned by an electrical company (59°21.231′N 18°6.550′E) as sampling location 2, and a gas station (59°21.376′N 18°6.313′E) as sampling location 3. The samples were collected at a depth of approximately 10 cm and were dried overnight at 25 °C. After drying, the samples were sieved to <2 mm using Sieve Shaker (AS200; Retsch, Haan, Germany) to remove stone and gravel. The samples were then stored in a refrigerator (4–6 °C) until chemical analysis.
Pressurized liquid extraction of samples
Approximately 40 mg of wood smoke particles or 1–1.5 g of soil was placed in an 11-mL stainless steel extraction cell and the IS (1-HP-d9) was added. The samples were extracted with methanol at 200 °C and 2000 psi in two 10-min extraction cycles, using an ASE 200 accelerated solvent extraction system (Dionex Corporation, Sunnyvale, CA, USA). The preheating time was 3 min, the equilibration time 9 min, purge with nitrogen in 1 min, and 30 % solvent flush. After extraction, the crude extracts were reduced in volume under a gentle stream of nitrogen in a TurboVap LV evaporator (Zymark Corporation, Hopkinton, MA, USA) at 65 °C to a final volume of approximately 0.5 mL. Method blanks, comprising micro-quartz fiber filters with added IS, were extracted, cleaned up, and analyzed in the same way.
Sample clean-up
Solid phase extraction (SPE) was used to clean up the extracts. The silica SPE cartridges (100 mg isolute; Biotage, Uppsala, Sweden) were conditioned with 3 mL hexane before 0.5 mL of the crude extract was added. The cartridges were washed with 0.5 mL hexane and the analytes were eluted with 3 mL methanol. The eluted fractions were evaporated to 0.5 mL and filtered by 0.2 μm Nylon filters (SUN-SRi, Rockwood, TN, USA) prior the analysis.
HPLC-MS/MS analysis
The analyses were performed on a HPLC system from Perkin Elmer (Waltham, USA) consisting of a Series 200 membrane degasser, two Series 200 micro pumps and a Series 200 autosampler. The chromatographic separation was performed on a C18 column (ACE 3, Ø 2.1 mm × 50 mm, 3 μm particle size, Advanced Chromatography Technologies, Aberdeen, Scotland). A C18 Guard-PAK™ HPLC pre-column (Waters, Millipore Corp., Milford, MA, USA) was used as a guard column prior the analytical column. Mobile phase A was 20 mM ammonium acetate and mobile phase B was acetonitrile. The loop volume was 5 μL and the mobile phase flow rate was set to 250 μL/min. The gradient condition used was: 0.1 min 30 % B, 2 min 60 % B, 4 min 60 % B, 6 min 90 % B, 7 min 90 % B, 7.1 min 30 % B. The system was equilibrated for 5 min with 30 % B before each run.
An Agilent Technologies 1100 series binary pump (Waldbronn, Germany) was used as a dopant introducing pump. The dopant flow rate was set to 25 μL/min.
A triple quadrupole mass spectrometer, PE SCIEX API 365 LC/MS/MS System (SCIEX Toronto, ON, Canada) with a PhotoSpray photoionization interface was used. The MS parameters were set as follows; curtain gas (N2): 8 arbitrary units (a.u.), nebulizer gas: 75 psi, collision gas (N2): 6 a.u., ion spray voltage: –3000 V, auxiliary gas: 8 a.u., temperature: 400 °C. The dwell time for each selected reaction monitoring (SRM) transition was set to 0.15 s. The optimal instrumental parameters for each analyte were obtained by tuning, using direct infusion of individual standard solutions. Quantitative analyses were performed in MS/MS mode. The identification of the analytes was based on comparison of SRM transitions and retention times with pure standard solutions. The analyte-specific parameters are shown in Table 2. Data were acquired and processed with Analyst software (ver. 1.4.2.; AB SCIEX, Concord, ON, Canada).
Extraction recoveries
Several parameters can affect the extraction efficiency; thus, experimental design was used as a first approach for screening for conditions with the highest impact on the extraction recoveries. Blank micro-quartz fiber filters were spiked with all the analytes in concentrations of approximately 10 μg prior the extraction, and IS was added to the crude extracts after the extraction. A two-level full factorial design (24) with 16 runs was constructed for the evaluation of four factors; temperature (100 °C or 200 °C), pressure (1000 psi or 2000 psi), number of extraction cycles (1 or 2), and the static time of each cycle (5 or 10 min). Each factor was given a minimum (–1) and a maximum (+1) value, the lowest setting getting the minimum value and vice versa, e.g., for the temperature, 100 °C was given –1 and 200 °C was given +1. The experimental design matrix and the outcome of each run (the average recovery of all analytes in duplicate) are shown in Table 3. The experimental data were processed using Modde software (ver. 10; MKS Umetrics, Umeå, Sweden).
Matrix effects
The matrix effects were determined by post-extraction standard addition experiments. The soil sample extracts were spiked with a standard solution containing all the analytes at concentration level of 0.1 μg/g soil. The wood smoke sample extracts were similarly spiked at a concentration level of 125 μg/g wood smoke particles. The response factors obtained by these additions were compared with those obtained for pure standard solutions, to evaluate the matrix effects.
Results and discussion
Chromatographic separation
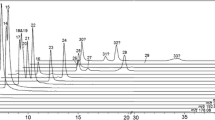
Example chromatograms for the analytes in a standard mixture are shown in Fig. 1. The analytes were separated within 7 min with a total run time of 12 min (including 5 min equilibration), and most of the peaks and all isomers were well resolved. It is particularly important with good chromatographic separation for the isomeric OH-PAHs, since they have the same precursor ions and transitions and cannot be differentiated by the mass spectrometer. Only 2-H-9-F could not be separated from the naphthol isomers; however, these compounds have different SRM transitions, thus avoiding errors in the quantification.

LC-MS/MS chromatograms of the quantitative ions for the analytes in a standard mixture (approximately 10 μg/mL)
Photoionization mass spectrometry
The general principles of APPI have been described in other studies [32, 33]. The ionization of the OH-PAHs was examined in both positive and negative modes. Ionization was, however, better in negative mode, by proton abstraction, because of the loss of the phenolic hydrogen, generating [M – H]– ions. Isomeric compounds gave the same precursor ions and transition and could not be differentiated without chromatographic separation. An ionization supporting dopant is usually introduced into the ionization region of APPI and the dopant is selected for its ability to undergo photoionization, requiring ionization energy (IE) below UV photon energy (10 eV). A composition of anisole (IE = 8.20) and toluene (IE = 8.83) 1:1 v/v was introduced as dopant directly into the heated nebulizer probe at a flow rate of 25 μL/min, together with the auxiliary gas, independently from the HPLC eluent flow. A higher signal and better ionization were achieved with this composition compared with others or when anisole and toluene were introduced separately. The dopant flow rate should be set to approximately 5 to 10 % of the eluent flow rate. The dopant molecules were ionized by the UV radiation, initiating ion-molecule reactions involving solvent molecules, which lead to formation of ionized analytes. The nebulizer gas pressure (recommended 70–80 psi), auxiliary and curtain gas flow (recommended 8–10 a.u.), and the vaporizer temperature (recommended 300–450 °C) were optimized in order to obtain good ionization. The fragmentation pattern was similar to fragmentation obtained in another study using ESI-MS/MS [41]. A loss of 28 Da (CO) was observed for all OH-PAHs when the precursor ions were fragmented. These fragments were the most abundant fragments for all OH-PAHs and were therefore chosen as identification (together with the retentions times) and quantification transitions. The fragmentation pattern for hydroxy biphenyls was different. A loss of 54 Da (C3H2O) and 76 Da (C6H4) was observed for 2-HBP and 4-HBP, respectively. The SRM transitions for each analyte are shown in Table 2.
Method performance
Calibration, detection limits, and precision
In order to study the linearity, six-point calibration plots were constructed for all the analytes in two concentration ranges; 1–50 ng/mL and 0.01–50 μg/mL. The coefficients of correlation, R2 were ≥ 0.997 in each case. The instrumental limits of detection (LOD) were calculated from signal-to-noise ratios (S/N) of standard solutions (obtained from Analyst software), using the definition S/N ratio greater than 3. The method limits of detection (MDL) and quantification (MQL) were defined as S/N > 3 and S/N > 10, respectively, and were determined by spiked sample extracts for analytes with background concentrations below MDL and MQL, whereas non-spiked sample extracts were used for analytes with background concentrations above the MDL and MQL. The instrumental LOD were in the range of 9–56 pg injected. The MDL for wood smoke and soil samples were 3–12 ng/g and 3–11 ng/g, respectively, whereas the MQL for wood smoke and soil samples were 5–38 ng/g and 6–36 ng/g, respectively, Table 4.
The instrumental LOD were higher compared with those obtained in some other studies [36, 42] (Table 5), which may depend on the fact that the instrument used in this present study belongs to an older generation of mass spectrometers. One can therefore assume that with a newer instrument equipped with APPI source, the instrumental LOD should be in the same range or even lower than those obtained in other studies, since APPI has been shown to be more sensitive compared with APCI and ESI [31, 32]. Instrumental LOD is often significantly lower than MDL, and does not reflect the practical detectability of the analytes in the matrix analyzed. It would therefore be more interesting to compare the MDL obtained in different studies. This information was not available in the literature for the analytes determined in this study. Lower LOD are desirable but not essential when analyzing matrices containing relatively high amounts of the target compounds, e.g., wood smoke, but are necessary when analyzing urine or water samples where they are present in much lower concentrations. The LOD and MQL were, however, sufficient for the sample matrices analyzed in this present study.
The precision and repeatability of the developed method was established through five independent sample preparations and analyses. Blank filters were spiked with a standard solution mixture prior to extraction and analyzed. The relative standard deviations (RSDs) for all analytes ranged from 2 to 12 %, Table 4.
Extraction recoveries
Results obtained from the experimental design were plotted in a coefficient plot showing that the main conditions affecting the extraction efficiency were the temperature and the pressure (p = 0.05), both having a significantly positive effect on the extraction recoveries, Fig. 2a. The coefficient plot displays the regression coefficients with confidence intervals and a coefficient is significant when the confidence interval does not cross zero. The temperature and the pressure were further plotted in a contour plot (Fig. 2b), showing that the recovery increased linearly with increased temperature and pressure and was highest at their maximum values (+1). Although there was no statistically significant evidence that the number of cycles and the static time of the cycles affect the extraction efficiency, when the results from the experimental design were plotted in a 4-D contour plot (Fig. 3), highest recovery was obtained when all of the screened parameters were set at their maximum values. Hence, the conditions for the extraction were chosen as follows: temperature 200 °C, pressure 2000 psi, 2× 10 min cycles.

(a) Coefficient plot of the coefficients that may affect the experimental response and (b) contour plot of the temperature and the pressure

4-D response contour of the recovery
The recoveries of the analytes ranged from 70 to 102 %, with RSDs lower than 3.3 % for all the compounds, Table 4. The extracts did not undergo any clean-up steps; therefore, it is assumed that the recoveries obtained only reflect the extraction step. It is important to stress that extraction of spiked filters and real samples differs because of analyte–matrix interactions for real samples. For this reason, wood smoke and soil samples were spiked with all the analytes and extracted with chosen extraction conditions, showing that the recoveries did not vary compared with the spiked filter samples. To the best of our knowledge, there are no certified reference materials (CRM) or certified methods provided by the National Institute of Standards and Technology (NIST) for determination of OH-PAHs in environmental matrices. The only CRM available are SRM 3672 and SRM 3673 “Organic contaminants in smokers’ urine” and “Organic contaminants in non-smokers’ urine,” respectively, which are not suitable to evaluate the method developed in the present study.
Sample clean-up
The SPE clean-up step was developed and improved by using standard solutions containing all of the analytes. After the SPE clean-up step, there was a loss of roughly 5 % of the analytes, yielding recoveries of 95–100 % in the standard solution. Losses in the evaporation and filtration steps were also examined, concluding that another 5 % of the analytes were lost in the evaporation step, whereas no losses occurred in the filtration step.
To determine the efficiency of the clean-up step, extracts spiked with all the analytes that were cleaned up on SPE cartridges were compared with extracts without undergoing the clean-up step. Yields ranging 92–104 % were achieved for the extracts from the SPE clean-up, whereas extracts that did not undergo the clean-up step suffered from signal suppressions and enhancements with yields ranging 85–120 %. It can therefore be stated that the SPE clean-up step is necessary to get rid of most of the matrix effects.
Matrix effects
Matrix effects occur when the signal responses of the analytes are suppressed or enhanced by the sample matrix. Ideally, when corresponding response factors for the spiked sample are compared with those obtained for pure standards, the relative response factors should be 100 %. Matrix effects between 92 and 104 % were observed for all the analytes in both matrices (Table 4), demonstrating that the matrix effects are small.
Application to samples
The developed method was applied to wood smoke particles and soil samples. Soil samples were prepared and analyzed in triplicate, whereas wood smoke samples were analyzed in duplicate. 1-HP was the only analyte detected in the soil samples, at sampling locations 2 and 3. Other OH-PAHs could not be detected above the LOD. Sampling location 2 contained 1-HP at a concentration level of 0.19 μg/g, whereas the concentration of 1-HP at sampling location 3 was 0.21 μg/g. Several OH-PAHs were detected in the wood smoke samples analyzed. Four analytes were detected in all wood smoke samples; 1-HP, 2-H-9-F, 2-HPh, and 3-HBaP, whereas 1-N was only detected in one wood smoke sample (sample 1). The concentrations in wood smoke sample 1, which was a HB case sample, ranged from 4.70 to 127 μg/g, and for wood smoke samples 2 and 3 (NB cases), from 0.88 to 84.8 μg/g and 1.43 to 97.4 μg/g, respectively. Chromatograms for the target analytes present in a wood smoke and a soil sample are shown in Fig. 4a and b. The determined concentrations of the analytes in the samples analyzed in this study and literature values are summarized in Table 6. There are few studies in the literature that have determined OH-PAHs in wood smoke and soil samples, and only a small number of OH-PAHs are determined, together with PAHs and/or oxy-PAHs [35, 36, 43, 44]. The different analytical methods reported in the literature for determination of OH-PAHs in smoke particles and soil samples are summarized in Table 7. GC-MS is the most used technique for detection of OH-PAHs in environmental samples. Compared with HPLC-MS, GC-MS is easier to work with since more parameters need to be adjusted and tuned when using HPLC-MS. However, a derivatization step is necessary when using GC-MS to convert OH-PAHs into thermostable and less polar compounds, and the run times are often longer compared to HPLC-MS methods. Various techniques, such as PLE, soxhlet, and ultrasonication are used for the extraction of OH-PAHs. While soxhlet is considered to be a solvent and a time-consuming procedure, PLE and ultrasonication are fast extraction techniques that work well for PAHs in general. In almost all studies, a clean-up step is used after the extraction to get rid of the polar compounds. This step is especially important when dealing with soil samples.

LC-MS/MS chromatograms of the quantitative ions for the analytes present in (a) wood smoke particles and (b) soil
There was no significant difference (p = 0.05) between the concentrations determined in the wood smoke samples in this study and the values obtained in the study by Cochran et al. [36], although the wood smoke samples in the latter study were collected from residential wood stoves and no further experiments on the combustion process were done. No literature values on the amount of 1-HP in soil could be found for comparison with soil samples analyzed in this present study.
Although wood smoke particles are assumed to be a primary source of OH-PAHs in the environment, OH-PAHs can be formed in soil by microbiological degradation of PAHs or end up in the soil as contaminants from secondary sources. That OH-PAHs are present in wood smoke samples suggests that they can be formed via other mechanisms than by microbiological and/or photochemical degradation of PAHs. The exact mechanism of OH-PAH formation during wood combustion is not fully clarified. It has, however, been suggested that it occurs through rapid radical reaction between hydroxyl radicals and PAHs because the combustion process occurs rapidly [43]. Thus, further investigations are required to fully understand the formation and occurrence of OH-PAHs in soil and wood smoke particles as well as in other environmental matrices.
Conclusions
A simple and fast method for analysis of OH-PAHs using PLE and HPLC-APPI-MS/MS was developed in this study. Simultaneous determination of 11 OH-PAHs with a run time of 12 min (including equilibration of 5 min) could be performed with high precision and low LOD and MQL. Lower LOD and MQL are expected with a newer mass spectrometer coupled to APPI source. A two-level full factorial experimental design was used for screening for conditions with the highest impact on the extraction. The extraction procedure was automated and suitable for a large number of samples. Extraction recoveries were good, and matrix effects were small. Five OH-PAHs were determined in wood smoke and soil samples in the concentration range 0.19–210 μg/g, using the developed method. Although OH-PAHs have gained more attention in recent years, a large part of the research done has focused on them as metabolites of PAHs. Their potential toxic and carcinogenic properties, together with few analytical methods reported in the literature for determination of OH-PAHs in environmental matrices, and the lack of CRM and certified methods shows that a significant amount of research is still needed in this area, as well as fast and simple methods for determination of these compounds in complex environmental matrices. The method developed in this study covers a broader range of different OH-PAHs, and the extraction, separation, and detection are specifically developed for OH-PAHs. As far as we know, OH-PAHs were determined in wood smoke particles and soil samples using APPI for the first time in this study. Accordingly, this study shows that HPLC-APPI-MS/MS can be a good option for determination of OH-PAHs in complex environmental matrices.
References
el-Bayoumy K (2008) Environmental carcinogens that may be involved in human breast cancer etiology. Chem Res Toxicol 5:585–590
Boffetta P, Jourenkova N, Gustavsson P (1997) Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Cause Control 8:444–472
Mastrangelo G, Fadda E, Marzia V (1996) Polycyclic aromatic hydrocarbons and cancer in man. Environ Health Perspect 104:1166–1170
Rantanen J (1983) Community and occupational studies of lung cancer and polycyclic organic matter. Environ Health Perspect 47:325–332
Phillips DH (1999) Polycyclic aromatic hydrocarbons in the diet. Mutat Res 443:139–147
Viau C, Hakizimana G, Bouchard M (2000) Indoor exposure to polycyclic aromatic hydrocarbons and carbon monoxide in traditional houses in Burundi. Int Arch Occup Environ Health 73:331–338
Vione D, Barra S, De Gennaro G, De Rienzo M, Gillardoni S, Perrone MG, Pozzoli L (2004) Polycyclic aromatic hydrocarbons in the atmosphere: monitoring, sources, sinks and fate. Ann Chim Rome 94:257–268
Boström CE, Gerde P, Hanberg A, Jernström B, Johansson C, Kyrklund T, Rannug A, Törnqvist M, Victorin K, Westerholm R (2002) Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ Health Perspect 110:451–488
Traczewska TM (2000) Changes of toxicological properties of biodegradation products of anthracene and phenanthrene. Water Sci Technol 41:31–38
Kazunga C, Aitken MD, Gold A, Sangaiah R (2001) Fluoranthene-2,3- and -1,5-diones are novel products from the bacterial transformation of fluoranthene. Environ Sci Technol 35:917–922
Chesis PL, Levin DE, Smith MT, Ernster L, Ames BN (1984) Mutagenicity of quinones: pathways of metabolic activation and detoxification. Proc Natl Acad Sci U S A 81:1696–1700
van Lipzig MMH, Vermeulen NPE, Gusinu R, Legler J, Frank H, Seidel A, Meerman JHN (2005) Formation of estrogenic metabolites of benzo[a]pyrene and chrysene by cytochrome P450 activity and their combined and supra-maximal estrogenic activity. Environ Toxicol Pharmacol 19:41–55
Kamiya M, Toriba A, Onoda Y, Kizu R, Hayakawa K (2005) Evaluation of estrogenic activities of hydroxylated polycyclic aromatic hydrocarbons in cigarette smoke condensate. Food Chem Toxicol 43:1017–1027
Hayakawa K, Onoda Y, Tachikawa C, Hosoi S, Yoshita M, Chung SW, Kizu R, Toriba A, Kameda T, Tang N (2007) Estrogenic/antiestrogenic activities of polycyclic aromatic hydrocarbons and their monohydroxylated derivatives by yeast two-hybrid assay. J Health Sci 53:562–570
Hirose T, Morito K, Kizu R, Toriba A, Hayakawa K, Ogawa S, Inoue S, Muramatsu M, Masamune Y (2001) Estrogenic/antiestrogenic activities of benzo[a]pyrene monohydroxy derivatives. J Health Sci 47:552–558
Jacob J, Seidel A (2002) Biomonitoring of polycyclic aromatic hydrocarbons in human urine. J Chromatogr B 778:31–47
Jongeneelen FJ (2001) Benchmark guideline for urinary 1-hydroxypyrene as biomarker of occupational exposure to polycyclic aromatic hydrocarbons. Ann Occup Hyg 45:3–13
Ferrari S, Mandel F, Berset JD (2002) Quantitative determination of 1-hydroxypyrene in bovine urine samples using high-performance liquid chromatography with fluorescence and mass spectrometric detection. Chemosphere 47:173–182
Hollender J, Koch B, Dott W (2000) Biomonitoring of environmental polycyclic aromatic hydrocarbon exposure by simultaneous measurement of urinary phenanthrene. pyrene and benzoa]pyrene hydroxides. J Chromatogr B 739:225–229
Kuusimäki L, Peltonen Y, Mutanen P, Peltonen K, Savela K (2004) Urinary hydroxy-metabolites of naphthalene, phenanthrene, and pyrene as markers of exposure to diesel exhaust. Int Arch Occup Environ Health 77:23–30
Schedl M, Wilharm G, Achatz S, Kettrup A, Niessner R, Knopp D (2001) Monitoring polycyclic aromatic hydrocarbon metabolites in human urine: extraction and purification with a sol-gel glass immunosorbent. Anal Chem 73:5669–5676
Gmeiner G, Krassnig C, Schmid E, Tausch H (1998) Fast screening method for the profile analysis of polycyclic aromatic hydrocarbon metabolites in urine using derivatization-solid-phase microextraction. J Chromatogr B 705:132–138
Gmeiner G, Gartner P, Krassnig C, Tausch H (2002) Identification of various urinary metabolites of fluorene using derivatization solid-phase microextraction. J Chromatogr B 766:209–218
Smith CJ, Walcott CJ, Huang WL, Maggio V, Grainger J, Patterson DG (2002) Determination of selected monohydroxy metabolites of 2-, 3-, and 4-ring polycyclic aromatic hydrocarbons in urine by solid-phase microextraction and isotope dilution gas chromatography–mass spectrometry. J Chromatogr B 778:157–164
Pigini D, Cialdella AM, Faranda P, Tranfo G (2006) Comparison between external and internal standard calibration in the validation of an analytical method for 1-hydroxypyrene in human urine by high-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 20:1013–1018
Li Y, Li AC, Shi H, Zhou S, Shou WZ, Jiang X, Naidong W, Lauterbach JH (2005) The use of chemical derivatization to enhance liquid chromatography/tandem mass spectrometric determination of 1-hydroxypyrene, a biomarker for polycyclic aromatic hydrocarbons in human urine. Rapid Commun Mass Spectrom 19:3331–3338
Galceran MT, Moyano E (1994) High-performance liquid chromatography-mass spectrometry (pneumatically assisted electrospray) of hydroxy polycyclic aromatic hydrocarbons. J Chromatogr A 683:9–19
Galceran MT, Moyano E (1996) Determination of hydroxy polycyclic aromatic hydrocarbons by liquid chromatography-mass spectrometry comparison of atmospheric pressure chemical ionization and electrospray. J Chromatogr A 731:75–84
Letzel T, Poschl U, Rosenberg E, Grasserbauer M, Niessner R (1999) In-source fragmentation of partially oxidized mono- and polycyclic aromatic hydrocarbons in atmospheric pressure chemical ionization mass spectrometry coupled to liquid chromatography. Rapid Commun Mass Spectrom 13:2456–2468
Letzel T, Poschl U, Wissiack R, Rosenberg E, Grasserbauer M, Niessner R (2001) Phenyl-modified reversed-phase liquid chromatography coupled to atmospheric pressure chemical ionization mass spectrometry: a universal method for the analysis of partially oxidized aromatic hydrocarbons. Anal Chem 73:1634–1645
Grosse S, Letzel T (2007) Liquid chromatography/atmospheric pressure ionization mass spectrometry with post-column liquid mixing for the efficient determination of partially oxidized polycyclic aromatic hydrocarbons. J Chromatogr A 1139:75–83
Robb DB, Covey TR, Bruins AP (2000) Atmospheric pressure photoionization: an ionization method for liquid chromatography-mass spectrometry. Anal Chem 72:3653–3659
Hanold KA, Fischer SM, Cormia PH, Miller CE, Syage JA (2004) Atmospheric pressure photoionization. 1. General properties for LC/MS. Anal Chem 76:2842–2851
Hutzler C, Luch A, Filser JG (2011) Analysis of carcinogenic polycyclic aromatic hydrocarbons in complex environmental mixtures by LC-APPI-MS/MS. Anal Chim Acta 702:218–224
Bandowe BAM, Bobocka J, Wilcke W (2011) Oxygen-containing polycyclic aromatic hydrocarbons (OPAHs) in urban soils of Bratislava, Slovakia: patterns, relation to PAHs and vertical distribution. Environ Pollut 159:539–549
Cochran RE, Dongari N, Jeong H, Beránek J, Haddadi S, Shipp J, Kubátová A (2013) Determination of polycyclic aromatic hydrocarbons and their oxy-, nitro-, and hydroxy-oxidation products. Anal Chim Acta 740:93–103
Wang G, Kawamura K, Zhao X, Li Q, Dai Z, Niu H (2007) Identification, abundance and seasonal variation of anthropogenic organic aerosols from a mega-city in China. Atmos Environ 41:407–416
Eriksson AC, Nordin EZ, Nyström R, Pettersson E, Swietlicki E, Bergvall C, Westerholm R, Boman C, Pagels J (2014) Particulate PAH emissions from residential biomass combustion: time-resolved analysis with aerosol mass spectrometry. Environ Sci Technol 48:7143–7150
Lyyranen J, Jokiniemi J, Kauppinen EI, Backman U, Vesala H (2004) Comparison of different dilution methods for measuring diesel particle emissions. Aerosol Sci Technol 38:12–23
Pettersson E, Boman C, Westerholm R, Boström D, Nordin A (2001) Stove performance and emission characteristics in residential wood log and pellet combustion, part 2: wood stove. Energy Fuels 25:315–323
Xu X, Zhang J, Zhang L, Liu W, Weisel CP (2004) Selective detection of monohydroxy metabolites of polycyclic aromatic hydrocarbons in urine using liquid chromatography/triple quadrupole tandem mass spectrometry. Rapid Commun Mass Spectrom 18:2299–2308
Pojana G, Marcomini A (2007) Determination of monohydroxylated metabolites of polycyclic aromatic hydrocarbons (OH-PAHs) from wastewater treatment plants. Int J Environ Anal Chem 87:627–636
Bi X, Simoneit BR, Sheng G, Fu J (2008) Characterization of molecular markers in smoke from residential coal combustion in China. Fuel 87:112–119
Simoneit BR, Bi X, Oros DR, Medeiros PM, Sheng G, Fu J (2007) Phenols and hydroxy-PAHs (arylphenols) as tracers for coal smoke particulate matter: source tests and ambient aerosol assessments. Environ Sci Technol 41:7294–7302
Acknowledgments
Ioannis Sadiktsis is acknowledged for the valuable comments on the manuscript and Hamid Ahmadi for skillful laboratory assistance. Robert Lindgren is acknowledged for his contributions to the wood combustion experiments. This study has been financed by Stockholm University and Umeå University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Avagyan, R., Nyström, R., Boman, C. et al. Determination of hydroxylated polycyclic aromatic hydrocarbons by HPLC-photoionization tandem mass spectrometry in wood smoke particles and soil samples. Anal Bioanal Chem 407, 4523–4534 (2015). https://doi.org/10.1007/s00216-015-8638-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8638-x