
Abstract
The analysis of paralytic shellfish toxins (PSTs) by liquid chromatography-mass spectrometry remains a challenge because of their high polarity, large number of analogues and the complex matrix in which they occur. Here we investigate the potential utility of high-field asymmetric waveform ion mobility spectrometry (FAIMS) as a gas-phase ion separation tool for analysis of PSTs by mass spectrometry. We investigate the separation of PSTs using FAIMS with two divergent goals: using FAIMS as a primary separation tool for rapid screening by electrospray ionization (ESI)-FAIMS-MS or combined with LC in a multidimensional LC-ESI-FAIMS-MS separation. First, a survey of the parameters that affect the sensitivity and selectivity of PST analysis by FAIMS was carried out using ESI-FAIMS-MS. In particular, the use of acetonitrile as a gas additive in the carrier gas flow offered good separation of all PST epimeric pairs. A second set of FAIMS conditions was also identified, which focussed PSTs to a relatively narrow CV range allowing development of an LC-ESI-FAIMS-MS method for analysis of PST toxins in complex mussel tissue extracts. The quantitative capabilities of this method were evaluated by analysing a PST containing mussel tissue matrix material. Results compared favourably with analysis by an established LC–post-column oxidation–fluorescence method with recoveries ranging from 70 to 106 %, although sensitivity was somewhat reduced. The current work represents the first successful separation of PST isomers using ion mobility and shows the promise of FAIMS as a tool for analysis of algal biotoxins in complex samples and outlines some critical requirements for its future improvement.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
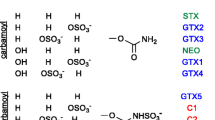
Paralytic shellfish toxins (PSTs) are potent neurotoxins produced by several marine dinoflagellates and freshwater cyanobacteria that are bio-accumulated by bivalves and lead to cases of paralytic shellfish poisoning (PSP) worldwide [1, 2]. The parent toxin of the class, saxitoxin, is a schedule 1 chemical weapon and has many structural analogues, which have been identified in algae and shellfish (Fig. 1). In most regulatory laboratories, the AOAC mouse bioassay has been used for routine analysis [3], but this method suffers from low sensitivity and poor reproducibility [4]. Recently, the replacement of mouse bioassays in Europe for the monitoring of lipophilic toxins [5] has led to the adoption/use of instrumental analytical methods, capable of more sensitive and reliable quantification as well as confirmation of the chemical nature of the detected toxins. The most commonly used chemical analytical methods for PSTs are those based on liquid chromatography (LC) coupled with fluorescence detection and using pre- or post-column oxidation reactions producing fluorescent PST derivatives [6–8]. These methods have some drawbacks related to system upkeep and lack of structural confirmation, which is only possible using mass spectrometry detection. In general, shellfish toxin analysis over the last several years has been moving towards the use of liquid chromatography coupled to tandem mass spectrometry detection (LC-MS/MS) with electrospray ionization (ESI), which has been successfully implemented for the regulatory monitoring of lipophilic shellfish toxins [9–11].

Structures of PSTs analysed
Compared with lipophilic classes of algal toxins, the analysis of PSTs by LC-MS remains a significant instrumental challenge for several reasons including (a) their highly polar structures limiting their retention in reverse phase LC, (b) the large number of structurally similar analogues including several epimeric pairs (Fig. 1), (c) labile precursor ions whose in-source fragments share m/z values with precursors or product ions of other analogues and (d) chemical interference and ionization suppression resulting from the complex shellfish tissue extract matrix. Despite these challenges, a number of LC-MS methods have been reported for the analysis of PSTs using hydrophilic interaction liquid chromatography (HILIC) mode of LC separation, which allows for retention and separation of highly polar PSTs [12–15]. This previous work has demonstrated the potential of the technique as well as some of its current limitations, including the presence of ESI signal suppression and matrix interferences [12].
High-field asymmetric waveform ion mobility spectrometry (FAIMS), also known as differential mobility spectrometry (DMS), is a relatively new mode of analytical separation that was developed for use with mass spectrometry by Guevremont et al. [16]. Since that time, several devices based on the same principles of separation have been made commercially available by MS instrument manufacturers [17–20]. The FAIMS device acts as a continuous ion filter to separate ions in the gas phase, at atmospheric pressure after they are produced by ESI but prior to entering the MS. The mechanism of separation in FAIMS is based on differences in the mobility characteristics of ions as they are subjected to an electric field oscillating between high and low field strengths [17, 21–25]. The main advantage of using FAIMS in analytical mass spectrometry lies in the orthogonality of the separation based on the size, charge, shape and polarisability of an ion, to the separations in both liquid chromatography and mass spectrometry. This orthogonality results in increased selectivity for isolation of a targeted analyte from interfering species, such as other isobaric analytes or matrix components. Thus, analytical MS methods incorporating a FAIMS filter step prior to MS analysis are able to selectively resolve and quantify species that otherwise cannot be selectively analysed by ESI-MS or LC-ESI-MS alone. When used as a primary separation tool, ESI-FAIMS-MS can provide sufficient selectivity to perform very rapid analyses (ca. 30 s/sample) of samples without the need for LC separation [24, 26]. For more complex samples that cannot be analysed by direct infusion electrospray, FAIMS can be combined with LC (LC-ESI-FAIMS-MS) to perform highly selective multidimensional separations [22].
The composition of the bath gas is a critical parameter for ion separation in FAIMS. Depending on the nature of the analyte and its interaction with the bath gas at high and low electric field strength, one of two behaviours can be observed [25]. Mixtures of helium and nitrogen are typically used to enhance decreases in mobility with increasing electric field referred to as P2 mode. This effect results from differences in the dynamics of interactions between analyte ions and the two gasses at high and low field strength [26]. Alternatively, CO2 and polar gas additives are used for studying increases in mobility at higher fields, referred to as P1 mode [17, 20, 27, 28]. This effect is related to clustering and declustering of analyte ions with polar gas molecules at low and high field strength, respectively [20]. Recently, attempts have been made to use an alternative mode of differential ion mobility, traveling wave ion mobility spectrometry (TWIMS), to separate PST isomers form one another [29]. This work showed that PST isomers could not be separated from one another as protonated or deprotonated ions using TWIMS. However, the authors were able to achieve epimers separation using TWIMS by analysing PST adducts with alkali earth metals in salt spiked solutions. In order to minimize matrix suppression for quantitative analysis, separation of protonated PSTs by ion mobility would be desirable.
The current work aims to evaluate FAIMS as a possible means of improving selectivity in LC-MS analysis of PSTs, where interference from matrix compounds has been identified as a limitation of current LC-MS methodology [12]. We begin by investigating the FAIMS separation of PST isomers from one another by direct ESI-FAIMS-MS using mixtures of either CO2 or He with N2, with the secondary goal of focussing PSTs to similar CV values for LC-ESI-FAIMS-MS. Recent literature has highlighted the significant benefits of polar solvent gas modifiers on differential mobility separations [17, 20, 27, 28]. Here, we investigate the impact of acetonitrile vapour on FAIMS separation of PSTs using a second FAIMS-MS system that has been modified for the addition of gas additives. An LC-ESI-FAIMS-MS method was then developed for analysis of PSTs in mussel tissue extracts. Finally, in order to evaluate the quantitative capabilities of this technique, a PST mussel tissue reference material was analysed, and results were compared with those obtained using LC-fluorescence detection (FLD) with post-column oxidation.
Experimental
Optima LC-MS-grade acetonitrile (CH3CN) was purchased from Fisher (Ottawa, ON, Canada). Ammonium formate (98 %) was purchased from Fisher Chemicals (Fairlawn, NJ, USA). Formic acid (98 %) was purchased from EMD (Darmstadt, Germany). Deionised water was produced by passing distilled water through a Milli-Q Gradient A10 deionised water system (Millipore, USA).
All PST standards and matrix materials were provided by the National Research Council Canada (Halifax, NS, Canada). These included the certified reference material (CRM) calibration solutions for the toxins in Fig. 1: CRM-STX-f, CRM-NEO-c, CRM-GTX2&3-c, CRM-GTX1&4-c and CRM-dcGTX2&3-b. Tissue matrix materials analysed included CRM-Zero-Mus, a mussel tissue homogenate certified to be free of PSTs as well as two different pilot-scale mussel tissue reference materials [30] whose PST concentrations, homogeneity and stability have been well characterised by an established analytical method, LC-pCox-FLD [8]. Extractions of the blended mussel tissues were prepared by using a modified version of a previously published extraction procedure [6]. Briefly, 4 g samples of mussel tissue homogenate was mixed with 4 mL of 0.1 M HCl and boiled for 10 min. After centrifugation, the supernatant was passed through a 60 mg OASIS HLB solid-phase extraction cartridge (Waters, Milford, MA), de-proteinated by adding one volume of acetonitrile and passed through a 4.5μm-pore-size filter. Quantification of PSTs in tissue reference material was carried out by spiking 100 μL of deionised water or mixed toxin standard into separate 400-μL aliquots of extract to construct a three-point standard addition calibration curve used to determine the concentration of the PSTs in the tissue.
ESI-FAIMS-MS and LC-ESI-FAIMS-MS on system 1
A Thermo Fisher LTQ-Orbitrap (Bremen, Germany) equipped with a Thermo Fisher FAIMS system was used for both infusion and LC-based analysis. ESI-FAIMS-MS was carried out by infusing toxin CRMs diluted to approximately 1–10 μM in 1:1 acetonitrile/water with 0.1 % acetic acid at 5 μL/min. MS source parameters for these infusions included a source temperature of 100 °C, capillary temp of 100 °C, capillary voltage of 10 V, a tube lens voltage of 40 V, spray voltage of +3500 V, sheath gas of 10, auxiliary gas of 5 and sweep gas of 0 (all arbitrary units). Mass spectral data were collected in full-scan mode from m/z 250 to 450 or in selected ion monitoring mode, all with an ion trap (IT) fill time of 100 ms, unless otherwise noted. FAIMS outer bias voltage was 10 V and gas flows, dispersion voltage and electrode temperatures are cited for individual experiments throughout. For flow-injection analysis and LC-ESI-FAIMS-MS experiments carried out at flow rates of 250 μL/min, harsher source parameters were used to ensure better desolvation prior to introduction into FAIMS. These included a source temperature of 300 °C, capillary temp of 350 °C, capillary voltage of 10 V, a tube lens voltage of 40 V, spray voltage of +3,500 V, sheath gas of 35, auxiliary gas of 30 and sweep gas of 0 (all arbitrary units).
Liquid chromatography was performed using an Agilent 1200 LC system with an Acquity UPLC 1.7 μm BEH Amide 2.1 × 100 mm column (Waters, Milford, MA, USA) operated in HILIC mode, similar to conditions reported previously [14]. LC flow rate was 250 μL/min, and the injection volume was 10 μL. The mobile phase used was 2 mM ammonium formate pH 3.5 (A) and 0.1 % formic acid in acetonitrile (B) using a linear gradient from 75 to 55 % B in 10 min, held for 2 min, before returning to the initial conditions.
ESI-FAIMS-MS investigation of gas additives on system 2
A FAIMS-equipped Thermo Fisher TSQ Quantum (San Jose, CA) was used for ESI-FAIMS-MS studies using gas modifiers. The standard setup was modified as described previously to enable the use of gas additives [20]. Briefly, a Waters UPLC system was used to deliver a reproducible flow of solvent into a mixing-T installed in-line with the buffer gas flow being delivered to the FAIMS electrodes. System 2 also used a modified electrode set having a 15 mm inner electrode and an improved desolvation region [18]. This resulted in a narrower electrode gap and overall higher electric field strength at a given applied dispersion voltage than system 1, which used a standard 13 mm inner electrode. A Harvard apparatus syringe pump was used to deliver PST standard solutions to the ESI needle tip at a flow rate of 15 μL/min. The ESI needle was operated in positive ion mode, and MS detection was carried out in selected reaction monitoring (SRM) scan mode using the conditions shown in Table 1. Optimised conditions for analysis of the mixture of PSTs in Fig. 1 included a dispersion voltage of 4,400 V, a gas flow of 4 L/min N2 with 1.25 % acetonitrile as the modifier and inner/outer electrode temperatures of 35/55 °C and CV values and SRM conditions in Table 1. Alternative FAIMS parameters for separation of specific epimeric pairs are described in figure captions and text in “Results and discussion”.
Results and discussion
Depending on the analytical method being developed, FAIMS optimization for PSTs was carried out towards one of two divergent goals. For selective direct analysis of PSTs by ESI-FAIMS-MS, the epimeric pairs GTX2/GTX3, GTX1/GTX4 and dcGTX2/dcGTX3 (Fig. 1) must be separated from one another since they are not resolved by mass spectrometry. Furthermore, the abundant product ions and in-source fragments of GTX2 and GTX3 are sulphur trioxide (SO3) loss product ions identical in composition to protonated NEO (Fig. 1), requiring complete separation of these compounds prior to MS analysis. On the other hand, in LC-ESI-FAIMS-MS, isolation of PSTs from interfering matrix components rather than from one another is the primary goal. The LC separation of PST epimers from one another is possible because of significant differences in molecular conformation [12, 31]. Since FAIMS acts as an ion filter, the number of simultaneous CV values that must be monitored at any one time during an LC separation must be minimised in order to minimise the impact on the overall duty cycle of the analysis. Thus, it becomes desirable to focus co-eluting analytes to the same CV values away from interfering matrix components, which have previously been identified as a limitation to LC-MS analysis of PSTs [12].
Separation of PSTs by FAIMS on system 1
The FAIMS separation properties of PSTs were investigated by infusing toxin standards at low flow rates for direct ESI-FAIMS-MS analysis. Each FAIMS parameter was investigated by scanning compensation voltage (CV), initially from −50 to 50 V. The parameters that were investigated included polarity and magnitude of dispersion voltage (DV), carrier gas composition and flow rate, electrode temperatures and ion trap fill time. An iterative approach was employed in optimizing where each parameter was first examined individually and then revisited after optimization of all other parameters in order to determine their cumulative effect.
Depending on the differential mobility behaviour of the analytes, they can be more effectively transmitted and/or separated through FAIMS with either positive or negative dispersion voltages, referred to as P2 and P1 mode, respectively [25]. Under all sets of conditions examined on system 1, PST transmission was at least 30-fold higher in P2 mode than in P1 mode, limiting the practical utility of the latter for PST separation. In P2 mode, a rapid decline in sensitivity was observed below the maximum dispersion voltage of −5,000 V, which was then used for all subsequent analyses.
Increased electrode temperature had the benefit of improving transmission of the non-sulphated PSTs, STX and NEO but had the undesirable effect of increasing fragmentation of the labile GTXs in the MS source. A decrease in electrode temperature also had a favourable impact on separation between GTX epimeric pairs, as can be seen in the selected ion compensation voltage (SICV) spectra in Fig. 2. The behaviour of α- and β-sulphated PST epimers during collisional induced dissociation has been well studied, and these compounds differ greatly in lability during collision-induced dissociation, both in the MS source and in MS/MS [12, 31]. This was exploited in order to observe partial PST separation by FAIMS on system 1 by monitoring the more labile epimer as its fragment ion formed in the source region. This approach gave a good approximation of the partial separation of epimers observed on system 1 and was used to investigate the effect of different parameters on FAIMS separation. However, it would not be appropriate for quantitative analysis of PSTs by ESI-MS as there is interference between the epimers in the form [M+H]+ of the labile epimer and [M+H-SO3]+ of the more stable epimer. In Fig. 2, SICV spectra of the m/z 396 [M+H]+ ion and its m/z 316 (SO3 loss) source fragment can be used as an approximation of the separation between the less labile GTX3 and the more labile GTX2, respectively. At high values of 70/90 °C inner/outer electrode temperature, little separation was observed between the epimers, but better separation is observed at the lowest value of 35/45 °C.

Extracted ion compensation voltage spectra showing the effect of electrode temperature on GTX2 (m/z 316) and GTX3 (m/z 396) separation at a 70/90 °C, b 50/70 °C and c 35/45 °C inner/outer electrode temperatures
At sample infusion flow rates, the FAIMS carrier gas flow rate did not have a significant impact on either sensitivity or epimer resolution. A gas flow rate of 3.5 L/min gave slightly improved sensitivity and separation compared with lower flow rates, while higher flow rates resulted in instability of the ion signal. Gas composition proved to have a significant impact on sensitivity and CV of transmission for PSTs but less impact on separation of epimeric pairs. In P2 mode, the addition of He to the N2 buffer gas resulted in a significant increase in signal intensity as shown in Fig. 3 for STX, and a shift to more negative CV values up to 50 % He, which is the maximum the instrument allows due to the risk of electrical discharge. This change in CV results from the shallow potential energy well-depth of the ion-He interaction when compared with N2 [32]. An alternative carrier gas modifier, CO2, has previously been reported to provide more gentle conditions for the detection of fragile ions as well as superior separation for small polar analytes in P1 mode [24, 26]. For PSTs, the addition of CO2 (from 0 to 20 % in N2) gave a modest improvement in sensitivity in P1 mode, but without enhancement of epimer separation. The relative intensity of the labile [M+H]+ ion of GTX 1 at m/z 412 did, however, increase with the addition of CO2 to the point where it was nearly equal in intensity with the SO3 loss in-source fragment ion at m/z 332, supporting a gentler introduction into the mass spectrometer than without CO2. However, compared with a buffer gas of 50 % He in N2, the absolute intensity was prohibitively low in P1 mode, even when CO2 was added.

Extracted ion compensation voltage spectra showing the impact of increasing %He in N2 for the analysis of protonated saxitoxin at m/z 300 by ESI-FAIMS-MS
The ion filtering effect of FAIMS significantly reduces the total ion current introduced into the mass spectrometer at any one time. This has a beneficial impact for ion trap mass analysers, which can be operated at much higher fill times than when all ions produced by ESI are introduced into the MS [33]. For PST toxin standards at 5 μM, ion trap fill times could be increased from the default value of 10 ms to above 1,000 ms without trap saturation. Provided the IT fill time is set to below the time spent at each CV step, it is possible to increase S/N in ESI-FAIMS-MS at no cost to analysis time or FAIMS resolution. Greater care must be taken in LC-ESI-FAIMS-MS experiments where achieving an acceptable number of data points across an LC peak becomes an important consideration.
ESI-FAIMS-MS/MS analysis of PSTs using gas additives on system 2
The recent FAIMS and DMS literature has been dominated by the use of polar solvent vapours as dopants in low percent amounts in the buffer gas to enhance selectivity of the FAIMS separation [17, 20, 27, 28]. This is believed to occur because of structure specific clustering/declustering between analyte ions and solvent molecules at low and high field strengths, respectively. We investigated the use of gas additives for PST analyses with a second ESI-FAIMS-MS system (system 2), which was modified to introduce a volatile solvent as an additive into the FAIMS gas flow. As reported previously for a broad range of other analytes [20], acetonitrile was found to have a favourable impact on both the sensitivity of PST analysis and on epimer separation in FAIMS. Without the use of acetonitrile, GTX1 and GTX4 showed no separation using 100 % N2 and were detected in P2 mode (DV = −4,000 V) on system 2, as shown in trace (a) of Fig. 4a. When 0.6 % acetonitrile was added to the carrier gas (trace (b) of Fig. 4a) baseline separation of the two epimers was observed in P1 mode (DV = +4,000 V), with improved sensitivity. The broad peak at CV = −41 V can be attributed to species transmitted through FAIMS as dimers or multimers at different CV values than the [M+H]+ ions detected at CV = −32 V (GTX1) and −27 V (GTX4), but which then decompose to monomers upon introduction into mass spectrometry. This assignment is supported by the fact that the CV = −41 V peak increases to 80 % relative intensity at higher analyte concentrations (8 μM). Using the more sensitive mass spectrometer in single reaction monitoring mode on system 2, toxin standards were analysed at lower analyte concentrations (as low as 0.1 μM) where the formation of multimeric species was minimised. The baseline separation of [M+H]+ ions of GTX1 and GTX4 epimers achieved using ESI-FAIMS-MS with 0.6 % acetonitrile as a gas additive at a dispersion voltage (DV) of 4,000 V allowed for the product ion spectra of each toxin to be acquired selectively without the need for liquid separation as shown in Fig. 4b and c. Since the behaviour of PSTs during collision induced dissociation has been well characterised [12, 30], these spectra serve as strong evidence that the CV peaks in Fig. 4 represent the true separation of the [M+H]+ ions of GTX1 and GTX4 epimers rather than of different gas phase conformations of a mixture of isomers resulting from solvation or protonation.

The effect of acetonitrile vapour dopant on the FAIMS separation of GTX1 and GTX4 using system 2. a 100 % N2 at DV = −4,000 V is shown in trace (a) and 0.6 % acetonitrile in N2 at DV = 4,000 V is shown in trace (b) using SRM parameters in Table 1. Product ion spectra of m/z 412 at a collision energy of 5 eV, collected using 0.6 % acetonitrile at CV = −32 V for GTX1 (b) and at CV = −26 V for GTX4 (c)
The temperature gradient between the inner and outer FAIMS electrodes was found to be a critical variable in the separation of PST epimers. The optimal 20 °C gradient for separation of GTX epimers used inner/outer electrode temperatures of 35/55 °C. When this gradient was decreased, the separation deteriorated. For example, GTX2 and GTX3, which were well separated at 20 °C difference, were only partially separated at 10 °C difference, and, with equal electrode temperatures, showed only minimal separation. However, the improved separation using an electrode temperature gradient was achieved at the cost of sensitivity of analysis of the sulphated PSTs with a decrease of between two- and tenfold, depending on the analogue. This is consistent with a previous study that showed that temperature differences between the inner and outer electrodes gave an increase in resolution and a decrease in sensitivity because of changes in the electric fields that are used for ion separation in FAIMS [34]. It was therefore not possible to achieve optimal separation and sensitivity for all PSTs under a single set of FAIMS conditions, and compromises were required for analysis of a mixture of PSTs. This involved choosing parameters that gave the best possible separation for the most difficult to separate epimeric pair but still gave adequate separation of each other pair. In general, improved separation of the GTX compounds at increased DV and percent acetonitrile dopant gave a broadening of the STX and NEO peaks and pushed them to higher CV values. Of the epimer pairs, GTX2 and GTX3 were the most difficult to separate from one another. Considering each of the epimeric pairs behaved similarly during CID in the vacuum of the mass spectrometer [31], these differences in separation between the different epimeric pairs can be attributed to differences in interactions with the buffer gas during the FAIMS separation. Figure 5 shows all PSTs analysed at DV = 4,400 V with 1.25 % acetonitrile additive, optimal conditions for GTX2 and GTX3 separation, which gave acceptable separation of all epimeric pairs. Under the conditions described in Table 1, [M+H]+ ions of all GTX epimer pairs are separated with a resolution of at least 0.6 (GTX2 and GTX3) and baseline resolution for other pairs. Minor peaks observed between CV = −45 and −60 V can be attributed to multimeric species of PST ions, which under these conditions are minimised compared with the protonated species. The more gentle conditions for introduction of labile GTX ions into MS afforded by the use of gas additives allowed for all species to be detected using their [M+H]+ ions as the parent ions in SRM mode.

a FAIMS separation of [M+H]+ ions of PSTs using 1.25 % acetonitrile as an additive in N2 gas at a dispersion voltage of 4,400 V with inner/outer electrode temperatures of 35/55 °C on system 2. b Expansion of the CV axis in (a) from −45 to −25 V. SRM detection conditions for each compound are given in Table 1
FAIMS optimization for LC-ESI-FAIMS-MS on system 1
It is evident that, for selective analysis of PSTs by direct ESI-FAIMS-MS(/MS), the use of gas additives is highly desirable as adequate separation between epimers was not otherwise achieved. However, other more significant challenges related to ionization exist that currently limit the possibility of doing direct analysis of PSTs in complex samples by this technique. In particular, the complex shellfish tissue extracts in which PSTs must be quantified cannot be analysed by direct electrospray ionisation due to severe ionization suppression from matrix components. Analysis of PSTs in these complex samples must instead be achieved using LC-ESI-FAIMS-MS. In this instrumental configuration, separation of PST epimers is not required as these can be readily separated by LC. Also, the CV switching time of FAIMS limits the number of different CV values that can be simultaneously monitored at any point in a chromatographic run. It therefore became desirable to focus PSTs to a narrower CV range but separated from potential matrix interference. The need to monitor multiple CVs simultaneously during an LC run also has a significant negative impact on sensitivity. For this purpose, the use of 50 % He in N2 carrier gas without gas additives was favoured as it gave the best sensitivity observed on system 1 while also transmitting the examined PSTs at a narrower range of CV values than observed with solvent gas additives.
In order to scale-up the MS source and FAIMS conditions from the 5 μL/min flow rate used for infusion to the 250 μL/min flow rate of the LC method, several parameters needed to be re-optimised. This was carried out with CV scans in single-ion monitoring mode for [M+H]+ and [M+H-SO3]+ ions using flow injection analysis of toxin standards. Initially, source parameters including spray voltage, temperature and capillary voltage needed to be optimised in order to achieve acceptable desolvation at the higher flow rate, but FAIMS parameters were observed to be dependent on flow rate and source parameters as well. In particular, the use of higher inner/outer electrode temperature (90/105 °C) resulted in roughly a fourfold increase in sensitivity at the higher flow rate. This temperature change caused an increase in-source fragmentation to the point where all sulphated toxins were much more sensitively detected as [M+H-SO3]+ source fragments than as [M+H]+ ions. Under these optimised high-flow conditions, little separation was observed between PST epimers as shown in Fig. 6. This has the added benefit of reducing the number of CVs that need to be monitored in an LC-ESI-FAIMS-MS experiment, which is important for sensitivity of the analysis. The additional peak detected in the dcGTX2/dcGTX3 standard was ruled out as being separated dcGTX3 by LC-ESI-FAIMS-MS, which showed that no toxin was detected at CV = −22.5 V and both epimers were detected at CV = −15.8 V. This peak can instead be assigned as a buffer or system contaminant which is effectively filtered by FAIMS during analysis of dcGTX2 and dcGTX3 at CV = −15.8 V. This type of separation of PSTs from isobaric chemical interference is the primary goal in using FAIMS in combination with LC.

CV optimization for transmission of PST standards through FAIMS on system 1 at 200 μL/min using flow injection analysis. STX, NEO, GTX3 and GTX4 were detected as their [M+H]+ ions at m/z 300, 316, 396 and 412, respectively, while GTX1, GTX2, dcGTX2 and dcGTX3 were detected as their [M+H-SO3]+ source fragments at m/z 332, 316, 273 and 273, respectively, all in single-ion monitoring mode. *Labelled peak at −21 V in dcGTX2/3 trace is a system contaminant separated from dcGTXs by FAIMS
The limiting factor for analytical sensitivity is the duty cycle of the FAIMS device, which has a switching time between different CV values of about 100 ms, the time required to empty the device of ions that experienced a particular CV. This means that limiting the number of CV values that must be monitored simultaneously is an effective way of limiting sensitivity losses observed when using FAIMS in combination with LC. Two approaches were investigated for limiting the number of monitored CVs. These included using time periods with a limited number of optimised CV values at a given retention time, or reducing the number of monitored CVs to three values, which provided coverage of all analytes close to, but not at their optimal CV. Table 2 shows optimised CV values for each analyte under the high flow conditions described in the previous sections, as well as their retention times in HILIC. There are three distinct groupings of PSTs by retention time in the HILIC separation, (a) GTX1 and GTX2 prior to 5 min, (b) GTX3 and GTX4 between 5 and 6.3 min and (c) STX and NEO after 6.3 min, while dcGTX2 and dcGTX3 span the first two periods eluting between 4.8 and 5.2 min. It was therefore possible to create a method where a maximum of 3 CV values were monitored at any one time as seen for PST standards in Fig. 7a and PSTs in a mussel tissue reference material in Fig. 7b. The primary limitation of this time-binned CV method was that small drifts in retention time over the course of an LC sequence were problematic, particularly the change at 6.3 min which either risked moving onto the front of the STX peak or the tail of the dcGTX3 peak.

LC-ESI-FAIMS-MS analysis of PSTs a in a mixed standard and b in mussel tissue extracts using retention time bins with optimised CV values for each analyte in single-ion monitoring mode as listed in Table 1
In order to evaluate the quantitative capabilities of LC-ESI-FAIMS-MS for the analysis of PSTs in mussel tissue, a more robust method needed to be developed that was not as sensitive to small variations in retention time, which are not uncommon in the analysis of tissue extracts by LC. For this purpose, close approximations to the maximum CV values of each toxin were used to limit the number of CV values which needed to be monitored to 3, but each of these was monitored continuously over the course of the analysis. These individual channels included GTX1-4 and NEO detected at CV = −17 V, dcGTX2 and dcGTX3 detected at CV = −14.5 and STX detected at CV = −19 V. This more robust method was less sensitive due to the fact that toxins were not detected at their optimum CV values but was more suitable for analysis of large sample sets where retention time could be expected to drift slightly.
The quantitative capabilities of LC-ESI-FAIMS-MS for the analysis of PSTs were evaluated by analysing a second mussel tissue reference material using a standard addition method and the continuous CV monitoring approach described above. This reference material consisted of a mixture of contaminated mussel tissue and toxin-containing algae previously produced on a pilot scale for the standardization of PST measurements [30]. Chromatograms from this analysis are shown in the Electronic supplementary material (ESM) compared with those for LC-MS analysis under the same conditions. From this comparison, the overall reduction in matrix species detected throughout the chromatographic run is evident, as well as some cases where chemical interference with PST peaks was filtered out by FAIMS (dcGTX2 and STX). In other cases, chemical interference affecting the detectability of PSTs was not effectively filtered by FAIMS at the present conditions (GTX4 and dcGTX3). For the most part, quantitative results from LC-ESI-FAIMS-MS agreed well with those from an established method for PST analysis, LC-post column oxidation-FLD [8]. The biggest limitation to this approach is the relatively poor sensitivity of the current mass spectrometer, which was not able to detect GTX4 or dcGTX3 in the reference material. This limitation applied equally in LC-MS and LC-FAIMS-MS experiments. In the future, both the selectivity and sensitivity of the analysis will be improved by use of a triple quadrupole instrument in SRM mode, which itself would be much more suitable for quantitative analysis in complex tissue extracts.
Conclusions
Our work has outlined the two ways in which FAIMS separation can be used for the analysis of PSTs, either by separation of PST analogues for direct ESI-FAIMS-MS or by focussing of analytes to a narrow CV range away from matrix interference for multidimensional LC-ESI-FAIMS-MS/MS analysis. For direct analysis, the use of low percent levels of acetonitrile (~1 %) as an additive in the carrier gas was necessary to achieve adequate separation of all PST epimeric pairs not resolved by mass spectrometry. The significant positive impact of gas additives is consistent with the recent literature in planar DMS devices [17, 28] as well as the cylindrical FAIMS device used here [20]. For LC-ESI-FAIMS-MS, 50 % He in N2 without the use of gas additives provided the best balance of sensitivity while limiting the number of CV values which needed to be simultaneously monitored at a given LC retention time. Faster CV switching time would allow for simultaneous monitoring of a larger number of CV values which would allow for the superior selectivity of the FAIMS device operated with gas additives to be exploited to a greater extent in the future. For improved analysis of highly labile compounds like PSTs by LC-ESI-FAIMS-MS, better desolvation without inducing in-source fragmentation than could be achieved here with system 1 would also be highly advantageous. This limitation was less evident with the alternative source design on system 2 and should have less of an impact going forward with modern MS instruments. These highlighted limitations represent practical aspects to the implementation of FAIMS technology rather than fundamental limitations of the technique, which we have shown here to be promising for the analysis of challenging classes of analytes, such as PSTs, in complex samples such as mussel tissue extracts. Future work will involve implementing LC-ESI-FAIMS-MS/MS with gas additives for improved selectivity and an investigation of direct PST quantification using ESI-FAIMS-MS/MS.
References
Vale P (2014) Saxitoxin and analogs: ecobiology, origin, chemistry, and detection. In: Botana LM (ed) Seafood and freshwater toxins: pharmacology physiology and detection, 3rd edn. CRC Press, Boca Raton
Quilliam MA (2003) The role of chromatography in the hunt for red tide toxins. J Chromatogr A 1000:527–548
MBA, AOAC 959.08 Official Methods of Analysis (2000) 17th edn. AOAC INTERNATIONAL, Gaithersburg, MD. Method 959.08
Hess P, Grune B, Anderson DA, Aune T, Botana LM, Caricato P, van Egmond HP, Halder M, Hall S, Lawrence JF, Moffat C, Poletti R, Richmond J, Rossini GP, Seamer C, Vilageliu JS (2006) Three Rs approaches in marine biotoxin testing. Altern Lab Anim 34:193–224
Anonymous (2011) Commission Regulation (EU) No 15/2011 of 10 January 2011 amending Regulation (EC) No 2074/2005 as regards recognised testing methods for detecting marine biotoxins in live bivalve molluscs. Official Journal of the European Union L 006 of 1 January 2011: 3–6
Anonymous (2005) AOAC official method 2005.06 quantitative determination of paralytic shellfish poisoning toxins in shellfish using pre-chromatographic oxidation and liquid chromatography with fluorescence detection. AOAC International, Gaithersburg
Rourke WA, Murphy CJ, Pitcher G, van de Riet JM, Burns BG, Thomas KM, Quilliam MA (2008) Rapid postcolumn methodology for determination of paralytic shellfish toxins in shellfish tissue. J AOAC Int 91:589–597
Van de Riet JM, Gibbs RS, Chou FW, Muggah FW, Rourke WA, Burns G, Thomas G, Quilliam MA (2009) Liquid chromatographic post-column oxidation method for analysis of paralytic shellfish toxins in mussels, clams, scallops and oysters: single-laboratory validation. J AOAC Int 92:1690–1704
Brana-Magdalena A, Leao-Martins JM, Glauner T, Gago-Martinez A (2014) Intralaboratory validation of a fast and sensitive UHPLC/MS/MS method with fast polarity switching for the analysis of lipophilic shellfish toxins. J AOAC Int 97:285–292
McNabb P, Selwood AI, Holland PT, Aasen J, Aune T, Eaglesham G, Hess P, Igarishi M, Quilliam M, Slattery D, Van de Riet J, Van Egmond H, Van den Top H, Yasumoto T (2005) Multiresidue method for determination of algal toxins in shellfish: single-laboratory validation and interlaboratory study. J AOAC Int 88(3):761–772
van den Top HJ, Gerssen A, McCarron P, van Egmond HP (2011) Quantitative determination of marine lipophilic toxins in mussels, oysters and cockles using liquid chromatography-mass spectrometry: inter-laboratory validation study. Food Addit Contam Part A 28(12):1745–1757
Dell’Aversano C, Hess P, Quilliam MA (2005) Hydrophilic interaction liquid chromatography-mass spectrometry for the analysis of paralytic shellfish poisoning (PSP) toxins. J Chromatogr A 1081:190–201
Zhou L, Yin Y, Fu W, Qui B, Lin, Yang Y, Zheng L, Li J, Chen G (2013) Determination of paralytic shellfish poisoning toxins by HILIC–MS/MS coupled with dispersive solid phase extraction. Food Chem 137:115–121
Blay P, Hui JPM, Chang J, Melanson JE (2011) Screening for multiple classes of marine biotoxins by liquid chromatography–high-resolution mass spectrometry. Anal Bioanal Chem 400:577–585
Watanabe R, Matsushima R, Harada T, Oikawa H, Murata M, Suzuki T (2013) Quantitative determination of paralytic sellfish toxins in cultured toxic algae by LC-MS/MS. Food Addit Contam Part A 8:1351–1357
Purves R, Guevremont R, Day S, Pipich CW, Matyjaszczyk S (1998) Mass spectrometric characterization of a high-field asymmetric waveform ion mobility spectrometer. Rev Sci Instrum 69:4094–4105
Schneider BB, Covey TR, Coy SL, Krylov EV, Nazarov EG (2010) Chemical effects in the separation process of a differential mobility/mass spectrometer system. Anal Chem 82:1867–1880
Prasad S, Belford M, Dunyach J, Purves R (2014) On an aerodynamic mechanism to enhance ion transmission and sensitivity of FAIMS for nano-electrospray ionization-mass spectrometry. J Am Soc Mass Spectrom 25:2143–2153
Shvartsburg AA, Smith RD, Wilks A, Koehl A, Ruiz-Alsonso D, Boyl B (2009) Ultrafast differential ion mobility spectrometry at extreme electric fields in multichannel microchips. Anal Chem 81:6489–6495
Purves RW, Ozog A, Ambrose SJ, Prasad S, Belford M, Dunyach J (2014) Using gas modifiers to significantly improve sensitivity and selectivity in a cylindrical FAIMS device. J Am Soc Mass Spectrom 25:1274–12844
Shvartsburg AA (2009) Differential ion mobility spectrometry. CRC Press, Boca Raton
Purves RW (2013) Enhancement of biological mass spectrometry by using separations based on changes in ion mobility (FAIMS and DMS). Anal Bioanal Chem 405:35–42
Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK (1993) A new method of separation of multi-atomic ions by mobility at atmospheric pressure using a high-frequency amplitude-asymmetric strong electric field. Int J Mass Spectrom Ion Process 128:143–148
Gabryelski W, Wu F, Froese KL (2008) Comparison of high-field asymmetric waveform ion mobility spectrometry with GC methods in analysis of haloacetic acids in drinking water. Anal Chem 75:2478–2486
Purves RW, Guevremont R (1999) Electrospray ionization high-field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal Chem 71:2346–2357
Ells B, Barnett DA, Purves RW, Guevremont R (2000) Detection of nine chlorinated and brominated haloacetic acids at part-per-trillion levels using ESI-FAIMS-MS. Anal Chem 72:5455–4559
Guevremont R, Purves RW, Barnett DA, Ells B (2006) FAIMS apparatus and method using carrier gases that contain a trace amount of a dopant species. US Patent no. 7,026,612
Rorrer LCIII, Yost RA (2011) Solvent vapour effects on planar high-field asymmetric waveform ion mobility spectrometry. Int J Mass Spectrom 300:173–181
Poyer S, Loutelier-Bourhis C, Mondeguer F, Enche J, Coadou G, Bossée A, Hess P, Afonso C (2014) Characterization of paralytic shellfish poisons by HILIC-IM-MS coupling. 62nd ASMS Conference on Mass Spectrometry and Allied Topics, Baltimore, MD
Reeves K, Thomas K, Quilliam MA (2004) Mussel tissue certified reference material for paralytic shellfish poisoning toxins. Proceedings of the 5th International Conference on Molluscan Shellfish Safety. Marine Institute, pg. 116–122. http://hdl.handle.net/10793/576. Accessed 23 May 2014
Dorr FA, Kovacevic B, Maksic ZB, Pinto E, Volmer DA (2011) Intriguing differences in the gas-phase dissociation behavior of protonated and deprotonated gonyautoxin epimers. J Am Soc Mass Spectrom 22:2011–2020
Barnett DA, Ells B, Guevremont R, Purves RW, Viehland LA (2000) Evaluation of carrier gases for use in high-field asymmetric waveform ion mobility spectrometry. J Am Soc Mass Spectrom 11:1125–1133
Hall AB, Coy SL, Kafle A, Glick J, Nazarov E, Vouros P (2013) Extending the dynamic range of the ion trap by differential mobility filtration. J Am Soc Mass Spectrom 24:1428–1436
Barnett DA, Belford M, Dunyach J, Purves RE (2007) Characterization of a temperature-controlled FAIMS system. J Am Soc Mass Spectrom 18:1653–1663
Acknowledgments
The authors gratefully acknowledge the technical assistance of Krista Thomas and Margaret McCooeye as well as the support and editorial assistance of Pearse McCarron and Michael Quilliam. The authors would like to thank Thermo Fisher for the loan of the TSQ Quantum.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 156 kb)
Rights and permissions
About this article

Cite this article
Beach, D.G., Melanson, J.E. & Purves, R.W. Analysis of paralytic shellfish toxins using high-field asymmetric waveform ion mobility spectrometry with liquid chromatography-mass spectrometry. Anal Bioanal Chem 407, 2473–2484 (2015). https://doi.org/10.1007/s00216-015-8488-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8488-6